
Синтез и свойства комплексов железа (II) и железа (III)
КУРСОВАЯ РАБОТА
по неорганической химии
СИНТЕЗ И СВОЙСТВА КОМПЛЕКСОВ ЖЕЛЕЗА (II) И ЖЕЛЕЗА (III)
Оглавление
Оглавление. 2
Введение. 3
1. Литературный обзор. 4
1.1 Теория кристаллического поля. 4
1.2 Мессбауэровская спектроскопия. 9
1.3 Комплексные соединения железа в растворе. 13
1.3.1 Гексацианоферраты(II, III). 14
1.3.2 Дитиосульфатоферрат (III). 18
1.3.3 Дифосфатоферрат. 18
1.3.4 Катион гексаамминжелеза (II). 18
1.3.5 Комплексы с другими лигандами. 19
1.3.6 Триоксалатоферраты (III, II) калия. 19
1.3.7 Хелаты. 20
1.3.7 Нитропруссид натрия. 22
2. Практическая часть. 25
2.1 Синтезы комплексов. 25
2.1.1 Синтез триоксалатоферрата (III) калия. 25
2.1.2 Синтез хелатного комплекса. 25
2.1.3 Выращивание кристалла берлинской лазури. 25
2.1.4 Выращивание кристалла турнбулевой сини. 26
Идентификация: 26
Выводы: 28
Список литературы. 29
Приложение. 31
Введение
Цель данной работы состоит в изучении строения и свойств комплексных соединений железа (II) и железа (III).
В ходе выполнения работы были поставлены следующие задачи:
1) изучение литературных данных о физических свойствах элементов VIIIB группы и их соединений, в частности, соединений железа;
2) анализ комплексных соединений железа (II) и железа (III) с различными лигандами с точки зрения теории кристаллического поля;
3) изучение литературных данных о строении цианидных комплексов железа (спектры Мессбауэра).
В ходе литературного поиска рассматриваются:
1) теория кристаллического поля;
2) эффект Мессбауэра;
3) комплексообразование в растворах.
В экспериментальной части предпринята попытка получения кристалла «берлинской лазури» - «турнбулевой сини», получены комплексы триоксалатоферрата(III) и хелатный комплекс.
1. Литературный обзор
1.1 Теория кристаллического поля
железо соединение лиганда кристаллическое поле
Для теоретического изучения комплексных соединений (КС) широко используется теория кристаллического поля (ТКП). Она была предложена Бетэ в 1929 г для кристаллов, а с 50-хх годов прошлого века стала широко использоваться в химии комплексных соединений (1).
Теория кристаллического поля исходит из того, что природа лигандов и их расположение вокруг центрального иона (симметрия комплекса) уменьшают вырождение d-орбиталей и изменяют их энергию. Рассмотрим это на примере комплексного иона октаэдрической симметрии (ML6)n+, в котором центральный атом имеет электронную конфигурацию d1. Ион M+ расположен в центре октаэдра, совпадающем с началом прямоугольной системы координат, а лиганды – в вершинах октаэдра.
Орбитали dx²–y² и dz² совпадают с координатными осями, а остальные три (dxy, dxz, dyz) проходят вдоль биссектрис соответствующих координатных углов. B отсутствие лигандов все пять орбиталей были энергетически равноценны. Но с появлением лигандов в вершинах октаэдра электроны, находящиеся на орбиталях dx²–y² и dz², испытывают сильное отталкивание от отрицательно заряженных лигандов или от отрицательного конца полярной молекулы. Другие три орбитали попадают в области с минимальными значениями отрицательного потенциала, поэтому вероятность нахождения электрона на орбиталях dxy, dxz, dyz будет больше. Это соответствует тому, что под действием лигандов прежде энергетически равноценные d-орбитали разделились на две группы: орбитали dx²–y² и dz²(dγ), энергетически невыгодные для электрона, и орбитали dxy, dxz, dyz(dε) с меньшей энергией. Схема расщепления d-орбиталей октаэдрическим (тетраэдрическим) окружением показана на рис. 1. Разность между dε и dγ-уровнями обозначается через Δокт (Δтетр) и называется параметром расщепления. В научной литературе орбитали обычно dγ и dε обозначают deg и dt2g, а параметр расщепления 10Dq (1).

рис 1. Диаграмма расщепления d-орбиталей в поле лигандов.
Из рис. 1 следует, что заселение любой dε-орбитали одним электроном приводит к уменьшению на 0,4Δокт энергии октаэдрического комплекса, т. е. стабилизирует его по сравнению со сферически симметричным ионом, а заселение электроном любой из dγ-орбиталей этот комплекс дестабилизирует на 0,6Δокт. В тетраэдрическом поле порядок расщепления d-орбиталей будет обратным, а потому энергия стабилизации на один электрон будет 0,6Δтетр, а дестабилизации - 0,4 Δтетр. Величина понижения энергии координационного соединения в результате перераспределения d-электронов по dε- и dγ-орбиталям называется энергией стабилизации кристаллическим полем (ЭСКП). Эта энергия зависит от числа электронов на dε- и dγ-орбиталях и вычисляется по формулам (1):
ЭСКП(окт) = (0,4n – 0,6m) Δокт
ЭСКП(тетр) = (0,6n – 0,4m) Δтетр
где – число электронов на нижнем подуровне, m – число электронов на верхнем подуровне. Параметр расщепления в октаэдрическом поле больше, чем в тетраэдрическом, содержащем те же лиганды, и равен Δокт = 9/4 Δтетр
Параметр расщепления Δ зависит от размеров центрального иона, его заряда, электронной конфигурации и от природы лиганда. Экспериментально его определяют по спектрам поглощения комплексных соединений. Возбуждение электрона с нижнего уровня на верхний сопровождается поглощением энергии и появлением в спектре полосы, максимум которой соответствует энергии расщепления Δ. Значение Δ обычно выражают в волновых числах ν = 1/λ см–1. Большинство значений Δ лежит в пределах о 10000 до 30000 см–1. (1 см–1 соответствует энергии E = hνc = 6,26∙10–34∙3∙1010∙1 = 2,0∙10–23 Дж = 11,96 Дж∙моль–1 = 1,25∙10–4 эВ) В ряду 3d-, 4d-, 5d-элементов при прочих равных условиях Δ увеличивается от периода к периоду на 30–35 %. Например, для (Co(NH3)6)3+Δ = 23000 см–1, для (Rh(NH3)6)3+Δ = 34000 см–1, для (Ir(NH3)6)3+Δ = 41000 см–1. Величина Δ возрастает при переходе от комплексов двухрядных ионов 3d-элементов к трехрядным. Так для (Fe(H2O)6)2+ и (Fe(H2O)6)3+ значения Δ равны соответственно 10400 см–1 и 13700 см–1. Из спектроскопических измерений была найдена последовательность расположения лигандов по возрастанию их влияния на величину расщепления Δ, называемая спектрохимическим рядом лигандов (2):
I- < Br- < SCN- « Cl- < F- < OH- « ONO- < C2O42- < OH2< NCS- < ЭДТА4- < Py « NH3 < En < NO2- < ДМГ < CN- < CO.
Некоторые лиганды (роданид, нитрит) имеют два варианта присоединения и потому два места в ряду.
В октаэдрических комплексах, образуемых ионами с электронными конфигурациями d4, d5, d6, d7, возможно различное размещение электронов – либо высоко-, либо низкоспиновое в зависимости от параметра расщепления Δ и энергии спаривания P. Последняя определяется как разность энергий межэлектронного взаимодействия низкоспиновой (НС) и высокоспиновой (ВС) конфигураций, деленная на число спаривающихся электронов, и приводится в справочниках. Очевидно, что низкоспиновое состояние реализуется тогда, когда P < Δ, а высокоспиновое – когда P > Δ. Сведения о некоторых свойствах комплексов d-элементов представлены в таблице 1(1).
Таблица 1.
| Электронная конфигурация координ. иона | Ион-комплексообразователь | P, см–1 | Лиганды | Δ, см–1 | Электр. конфигурация октаэдр. иона | Спиновое состояние |
d4 | Cr2+ | 23500 | H2O | 13900 |
| BC |
Mn3+ | 28000 | H2O | 21000 |
| BC | |
d5 | Mn2+ | 25200 | H2O | 7800 |
| BC |
Fe3+ | 30000 | H2O | 13700 |
| BC | |
d6 | Fe2+ | 17700 | H2O | 10400 |
| BC |
| 17700 | CN– | 33000 |
| HC | ||
Co3+ | 21000 | F– | 1300 |
| BC | |
| 21000 | NH3 | 23000 |
| HC | ||
d7 | Co2+ | 22500 | H2O | 10100 |
| BC |
В рамках ТКП высокоспиновый комплекс (Fe(H2O)6)2+ с электронной конфигурацией ![]() будет менее устойчив (ЭСКП = 0,4Δокт), чем низкоспиновый (Fe(H2O)6)3+ (электронная конфигурация
будет менее устойчив (ЭСКП = 0,4Δокт), чем низкоспиновый (Fe(H2O)6)3+ (электронная конфигурация ![]() ЭСКП = 2,4 Δокт).
ЭСКП = 2,4 Δокт).
Распределение электронов между нижним (dγ) и верхним (dε) уровнями в тетраэдрических комплексах также зависит от соотношения Δ и P, но поскольку Δтетр < Δокт, тетраэдрические комплексы обычно остаются высокоспиновыми.
В отличие от метода валентных связей, ТКП, основываясь на электронной конфигурации центрального атома, положении лигандов в спектрохимическом ряду и симметрии комплекса, позволяет не только объяснять, но и предсказывать магнитные и спектроскопические свойства комплексов.
С физической точки зрения ТКП является весьма приближенной, поскольку учитывает только электростатическое взаимодействие между комплексообразователем и лигандами. ТКП не дает объяснения устойчивости комплексов с электронными конфигурациями центрального атома d0 и d10, однако существование подобных комплексов легко объяснимо с позиций метода молекулярных орбиталей.
Таблица 2. Физические свойства элементов триады железа (2).
| Свойства | 26Fe | 27Co | 28Ni |
| Атомная масса | 55,85 | 58,93 | 58,70 |
| Электронная конфигурация | (Ar)3d64s2 | (Ar)3d74s2 | (Ar)3d84s2 |
| 0,126 | 0,130 | 0,124 |
| 0,08 | 0,08 | 0,079 |
Энергия ионизации Э0 → Э+ , эВ | 0,58 | 0,94 | 1,28 |
| Возможные степени окисления | +2, +3, +6 | +2, +3 | +2, +3, +4 |
| кларк, ат.% (распространенность в природе) | 1,5 | 1∙10-3 | 3∙10-3 |
| Агрегатное состояние (н. у.) | Т В Е Р Д Ы Е В Е Щ Е С Т В А | ||
| Цвет | серебристо-серый | серо-стальной | серебристо-белый |
tпл, 0С | 1539 | 1493 | 1455 |
Tкип, 0С | 3070 | 2880 | 2800 |
Плотность d, г/см3 | 7,87 | 8,9 | 8,91 |
Стандартный электродный потенциал | -0,440 | -0,277 | -0,250 |
| Фото |
|
|
|



1.2 Мессбауэровская спектроскопия
Мессбауэровская спектроскопия (гаммарезонансная спектроскопия), основана на явлении излучения и резонансного поглощения γ-квантов атомными ядрами в твердых телах без потери части энергии на отдачу ядра. При этом внутренняя энергия решетки твердого тела не изменяется (не происходит возбуждения фотонов - колебательных квантов). Это явление названо эффектом Мессбауэра. Эффект Мессбауэра позволяет наблюдать ядерное резонансное поглощение (рассеяние) со спектральными линиями естественной ширины Г, которая обычно лежит в интервале от 10-9 до 10-5 эВ, что соответствует временам жизни первых возбужденных (так называемых мессбауэровских) ядерных уровней 10-6 ≥ t ≥10-10с. Резонансное поглощение γ -квантов возможно лишь при Е0 = Е'0 (где Е0 и Е'0 - энергии возбужденных состояний излучающего и поглощающего ядер соответственно) (3) (рис. 2).
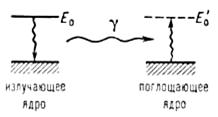
Рис. 2. Схематическое изображение процессов излучения и резонансного поглощения γ-квантов.
Для наблюдения спектра поглощения необходимо контролируемым образом изменить энергию γ-квантов DЕ и найти зависимость интенсивности прошедшего через поглотитель пучка γ-квантов как функцию этого изменения. Наиболее удобный и обычно применяемый способ - доплеровское изменение энергии DЕД, возникающее при перемещении источника излучения (или поглотителя) с варьируемой скоростью u. Тогда DEД = E0u/c (с-скорость света). Величины электрон-ядерных взаимодействий, обусловливающих различие Е0 и Е'0 для одинаковых нуклидов, соответствуют диапазону u в интервале — 10 см/с ≤ u ≤ 10 см/с и обычно составляют менее 10-6 эВ. Измеряя интенсивность прошедшего через поглотитель γ-излучения как функцию скорости u, получают мессбауэровской спектр, характеристиками которого являются положение линий в шкале скоростей, их число, относительная интенсивность, форма и площадь. Для измерения зависимости резонансного поглощения от u используют мессбауэровской спектрометр, упрощенная схема которого представлена на рис. 3 (4). Все нерезонансные процессы поглощения γ -квантов в веществе от u не зависят. Естественно, что в случае наличия различных изотопов в источнике излучения и поглотителе невозможно компенсировать различие Е0 и Е'0, которое, как правило, более 10 эВ и обусловлено не электрон-ядерными взаимодействиями, а различиями в ядерном строении. Таким образом, мессбауэровская спектроскопия обладает свойством абсолютной избирательности: резонансное поглощение возможно лишь в случае, когда в источнике излучения и поглотителе существуют ядра одного и того же изотопа (в возбужденном и основном состояниях соответственно). Другие элементы и изотопы не оказывают на него влияние. Количество спектральных линий поглощения и их положение в энергетической шкале зависят от значений спинов ядер в основном и возбужденном состояниях и природы электрон-ядерных взаимодействий в данном веществе, наличия внутриатомных магнитных полей, градиентов электрических полей, природы химической связи.
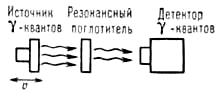
Рис. 3. Упрощенная схема мессбауэровского спектрометра; источник γ-квантов с помощью механического или электродинамического устройства приводится в возвратно-поступательное движение со скоростью u относительно поглотителя. С помощью детектора измеряется зависимость интенсивности потока γ-квантов, прошедшего через поглотитель от скорости (3).
Эффект Мессбауэра открыт Р. Мессбауэром в 1958 г., в 1961 г. за это открытие автор удостоен Нобелевской премии.
Магнитное дипольное взаимодействие обычно наблюдается в магнитоупорядоченных веществах (ферро-, антиферро-, ферримагнетиках), в которых на ядра действуют сильные магнитные поля от электронных оболочек. Оно приводит к расщеплению основного и возбужденного состояний ядер, в результате чего в спектре поглощения появляется несколько спектральных линий, число которых определяется величинами спинов ядер в этих состояниях и правилами отбора (например, для ядра 57Fe равно 6).
Энергия магнитного дипольного взаимодействия пропорциональна произведению напряженности магнитного поля (Н) на ядре на магнитный момент m ядра. Измерение (Н) дает возможность изучать электронное и спиновое строение исследуемого соединения и релаксационные эффекты.
Магнитное дипольное взаимодействие широко используется для изучения электронной и спиновой структуры химических соединений (высокоспиновые, низкоспиновые соединения), при исследовании магнитных свойств вещества в зависимости от характера химических связей. Важной особенностью мессбауэровской спектроскопии при изучении магнитных дипольных взаимодействий является высокая чувствительность спектров к локальному окружению мессбауэровских атомов. Поэтому эта область развита для изучения металлов, сплавов, твердых растворов, включая вопросы исследования фазового состава, дефектности, фазовых переходов, упорядочения (3).
 А
А
 Б
Б
Рис. 4. Температурная зависимость магнитного момента комплекса (Fe(RSalen)2)+PF6- (А) и мессбауэровские спектры комплекса Fe при температурах 293, 125 и 4,2 К (Б)(5).

Уникальная информативность мессбауэровской спектроскопии, относительная простота эксперимента и разработанные теоретические основы обусловили широкое применение мессбауэровской спектроскопии в физике и химии твердого тела, ядерной физике, геологии и археологии, аналитической химии, химической технологии.
1.3 Комплексные соединения железа в растворе
Железо, будучи переходным элементом, является типичным комплексообразователем. Железо образует устойчивые КС, находясь в валентном состоянии Fe (III) и Fe (II). Комплексообразование стабилизирует соединения малоустойчивой для железа степени окисления +2.
Железо образует достаточно большое количество комплексных соединений. Наиболее характерным для железа (II) и железа (III) является координационное число (КЧ) = 6 (реже 4 и 5). Способность к комплексообразованию более характерна для железа в С/О = +3. Рассмотрим некоторые комплексы железа и методы их получения.
1.3.1 Гексацианоферраты(II, III)
Для железа (II) очень устойчивы цианидные комплексы (цианоферраты (II)). Наиболее известным из них является гексацианоферрат (II) калия — желтая кровяная соль.
Желтая кровяная соль (гексацианоферрат (II) калия — K4(Fe(CN)6)) известна с середины XVIII века. Первоначально ее получали сплавлением отходов скотобоен (например, крови животных, копыт, кож и т. п.) с поташем (К2СO3) и железными обрезками. После остывания расплава и выщелачивания его водой получали желтую кровяную соль.
Сейчас гексацианоферрат (II) калия получают, действуя избытком KCN на соли двухвалентного железа:
FeCl2 + 6KCN = K4(Fe(CN)6) + 2KC1.
При добавлении к раствору солей трехвалентного железа раствора желтой кровяной соли образуется темно-синий осадок, называемый берлинской или npyccкой лазурью (13):
4FeCl3 + 3K4(Fe(CN)6) = FeIII4 (FeII(CN)6)3 + 12KCl.

Фото. 2. Берлинская лазурь
Это — качественная реакция на соли Fe3+.
Если на желтую кровяную соль подействовать окислителем (хлором, перманганатом калия), то получается комплексная соль трехвалентного железа гексацианоферрат (III) калия — K3(Fe(CN)6), которая окрашена в красный цвет и называется красной кровяной солью.
2K4(Fe(CN)6) + С12 = 2K3(Fe(CN)6) + 2KC1.

Фото. 1. Красная кровяная соль.
Красная кровяная соль (комплекс (Fe(CN)6)3-), менее устойчива, чем желтая (комплекс (Fe(CN6))4-), и поэтому очень ядовита.
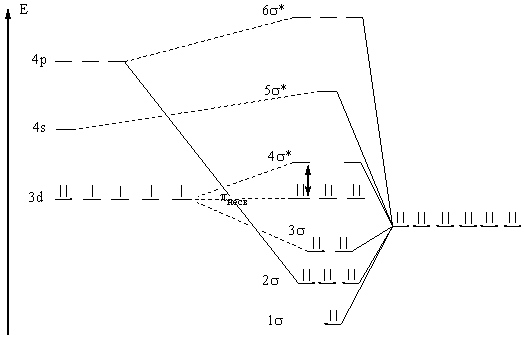
Рис. 5. Энергетическая диаграмма (Fe(CN6))4- (метод МО).
При взаимодействии растворов солей двухвалентного железа с красной кровяной солью образуется темно-синий осадок, называемый турнбулевой синью:
3FeCl2 + 4K3(Fe(CN)6) = FeIII4 (FeII(CN)6)3 + 6KC1 + 6KCN.
Это — качественная реакция на соли Fe2+.
Ранее считалось, что при этом образуется гексацианоферрат (III) железа (II), то есть FeII3(Fe(CN)6)2, именно такую формулу предлагали для «турнбулевой сини». Теперь благодаря исследованиям по методу мессбауэровской спектроскопии известно, что турнбулева синь и берлинская лазурь — одно и то же вещество, а в процессе реакции происходит переход электронов от ионов Fe2+ к гексацианоферрат (III)- иону (6):
Fe2+ + (Fe3+(CN)6)3- → Fe3+ + (Fe2+(CN)6)4-.
Этот процесс происходит практически мгновенно, а обратную реакцию можно осуществить лишь в вакууме при 300 0С (6). Этот факт объясняется, по-видимому, тем, что комплекс (FeII(CN)6)4- более устойчив, чем (FeIII(CN)6)3-.
Турнбулева синь и берлинская лазурь плохо растворимы в воде, что затрудняет их использование в виде красок. Для повышения растворимости гексацианоферратов вводят ионы калия вплоть до состава KFe(Fe(CN)6) (последнее соединение называется растворимой берлинской лазурью). По мере увеличения содержания ионов калия цвет соединений меняется от темно- до светло-синего.
Схема структуры растворимой «берлинской лазури» – «турнбулевой сини» (кристаллогидрат вида KFeIII(FeII(CN)6)·H2O) приведена на рисунке 6 (6).

Рис. 6. Кристаллическая решетка «берлинской лазури»
Из неё видно, что атомы Fe2+ и Fe3+ располагаются в кристаллической решётке однотипно, однако по отношению к цианидным группам они неравноценны, преобладает тенденция к размещению Fe2+ между атомами углерода, Fe3+ - между атомами азота, ионы К+ и молекулы H2O располагаются в пустотах кристаллической решетки.
Хотя состав, структура и кристаллические решетки берлинской лазури и турнбулевой сини идентичны, различные исторические названия продолжают сохраняться, отражая химию ионов железа в различных степенях окисления.
1.3.2 Дитиосульфатоферрат (III)
Это комплексное соединения образуется при взаимодействии железа 3+ с тиосульфатом натрия. Комплексное соединение окрашено в интенсивный фиолетовый цвет (8).
FeCl3+ 2 Na2S2O3 → 3 NaCl + Na(Fe(S2O3)2)
Далее окраска исчезает вследствие восстановления данной соли до бесцветной соли железа (II) тиосульфата и тетратионата (9).
2Na(Fe(S2O3)2) →FeS2O3+ FeS4O6+Na2S2O3
1.3.3 Дифосфатоферрат
В кислой среде фосфаты и фосфорная кислота с ионами железа образуют бесцветные комплексы (Fe(PO4)2) 3-(12):
FeCl3 + H3PO4 + K3PO4 → K3(Fe(PO4)2) + 3HCl
1.3.4 Катион гексаамминжелеза (II)
Катион (Fe(NH3)6)2+ малоустойчив и водой мгновенно разлагается (14):
(Fe(NH3)6)2+ + 6H2O = Fe(OH)2↓ + 4NH3∙H2O + 2NH4+
1.3.5 Комплексы с другими лигандами
К числу высокоспиновых комплексов Fe2+, образованных монодентантными лигандами, относятся акваионы (Fe(H2O)6)2+, КС с галогенид-ионами, например, K2(FeF4), в структуре которого зафиксированы конденсированные октаэдры (FeF6) (12).
Железо (II) в КС с полидентантными лигандами неустойчиво, так как быстро окисляется кислородом воздуха и водой, переходя в более термодинамически стабильные КС железа (III) (10).
1.3.6 Триоксалатоферраты (III, II) калия
Триоксалатоферрат (III) калия получают при взаимодействии сульфата железа (III), оксалата бария, и оксалата калия в водном растворе:
Fe2(SO4)3 + Ba2(C2O4)2 + 4K2C2O4 = 2K3(Fe(C2O4)3) + 2BaSO4 + K2SO4
Под действием света с длиной волны меньше 490 нм Fe(III) количественно восстанавливается до Fe(II) за счет окисления части ионов C2O42- до CO2 (14):
2K3(Fe(C2O4)3) = 2K2(Fe(C2O4)2) + K2C2O4 + 2CO2 ↑
Образующийся при этом оксалатный комплекс железа (II) хорошо растворим в воде, и выделяется из раствора в виде кристаллогидрата K2(Fe(C2O4)2) ∙6H2O золотисто-желтого цвета.

Фото. 3. Триоксалатоферрат (II) калия
1.3.7 Хелаты
Термин хелат (англ. chelate от греческого «клешня») принят для обозначения циклических структур, которые образуются в результате присоединения катиона к двум или более донорным атомам, принадлежащим одной молекуле комплексона. В соответствии с термином хелат эти соединения следует представлять в виде какого-то краба, который своими полидентатными клешнями прочно захватывает ион металла, и чем больше клешней, тем прочнее захват. Как буквальный перевод слова chelate в литературе до сравнительно недавнего времени для обозначе