
Дыхание растений
Дыхание — процесс универсальный. Оно является неотъемлемым свойством всех организмов, населяющих нашу планету, и присуще любому органу, любой ткани, каждой клетке, которые дышат на протяжении всей своей жизнедеятельности. Дыхание всегда связано с жизнью, тогда как прекращение дыхания — с гибелью живого.
Жизнь организма в целом, как и каждое проявление жизнедеятельности, необходимо связаны с расходованием энергии. Клеточное деление, рост, развитие и размножение, поглощение и передвижение воды и питательных веществ, разнообразные синтезы и все другие процессы и функции осуществимы лишь при постоянном удовлетворении обусловленных ими потребностей в энергии и пластических веществах, которые служат клетке строительным материалом.
Источником энергии для живой клетки служит химическая (свободная) энергия потребляемых ею питательных веществ. Распад этих веществ, происходящий в акте дыхания, сопровождается освобождением энергии, которая и обеспечивает удовлетворение жизненных потребностей организма.
Сам же процесс дыхания представляет собой сложную многозвенную систему сопряженных окислительно-восстановительных процессов, в ходе которых имеет место изменение химической природы органических соединений и использование содержащейся в них энергии.
1.Дыхание. Определение. Уравнение. Значение дыхания в жизни растительного организма. Специфика дыхания у растений
Клеточное дыхание — это окислительный, с участием кислорода распад органических питательных веществ, сопровождающийся образованием химически активных метаболитов и освобождением энергии, которые используются клетками для процессов жизнедеятельности.
Суммарное уравнение процесса дыхания:
С6Н12О6 + 602 ► 6С02 + 6Н20 + 2875 кДж/моль
Не вся энергия, высвобождаемая при дыхании, может быть использована в процессах жизнедеятельности. Используется организмом в основном та энергия, которая аккумулируется в АТФ. Синтезу АТФ во многих случаях предшествует образование разности электрических зарядов на мембране, что, в свою очередь, связано с разностью концентраций ионов водорода по разные стороны от мембраны. Согласно современным представлениям, е только АТФ, но и протонный градиент служат источником энергии для различных процессов жизнедеятельности клетки. Обе формы энергии могут быть использованы на процессы синтеза, процессы поступления, передвижения питательных веществ и воды, создание разности потенциалов между цитоплазмой и внешней средой. Энергия, не накопленная в протонном градиенте и АТФ, в основном рассеивается в виде тепла или света и является для растения бесполезной.
Значение дыхания в жизни растения.
Дыхание — один из центральных процессов обмена веществ растительного организма. Выделяющаяся при дыхании энергия тратится как на процессы роста, так и нa поддержание в активном состоянии уже закончивших рост органов растения. Вместе с тем значение дыхания не ограничивается тем, что это процесс, поставляющий энергию. Дыхание, подобно фотосинтезу, сложный окислительно_восстановительный процесc, идущий через ряд этапов. На его промежуточных стадиях образуются органические соединения, которые затем используются в различных метаболических реакциях. К промежуточным соединениям относят органические кислоты и пентозы образующиеся при разных путях дыхательного распада. Таким образом, процесс дыхания — источник многих метаболитов. Несмотря на то что процесс дыхания в суммарном виде противоположен фотосинтезу, в некоторых случаях они могут дополнять друг друга. Оба процесса являются поставщиками как энергетических эквивалентов (АТФ, НАДФ-Н), так и метаболитов. Как видно из суммарного уравнения, в процессе дыхания образуется также вода. Эта вода в крайних условиях обезвоживания может быть использована растением и предохранить его от гибели. В некоторых случаях, когда энергия дыхания выделяется в виде тепла, дыхание ведет к бесполезной потере сухого вещества. В этой связи при рассмотрении процесса дыхания надо помнить, что не всегда усиление процесса дыхания является полезным для растительного организма.
2. Основные этапы становления учения о дыхании растений
Научные основы учения о роли кислорода в дыхании были заложены трудами А.Л.Лавуазье. В 1774 г. кислород независимо открыли Пристли и Шееле, а Лавуазье дал название этому элементу. Изучая одновременно процесс дыхания животных и горение, Лавувзье в 1773-1783 гг. пришел к выводу, что при дыхании, как и при горении, поглощается кислород и образуется углекислый газ, причем в том и другом случаях выделяется тепло. На основании своих опытов он заключил, что процесс горения состоит в присоединении кислорода к субстрату и что дыхание есть медленно текущее горение питательных веществ в живом организме.
Я.Ингенхауз в 1778-1780 гг. показал, что зеленые растения в темноте, а незеленые части растений и в темноте, и на свету поглощают кислород и выделяют углекислый газ. В своей работе, опубликованной в 1779 г. он писал:
«Когда солнце, поднявшееся над горизонтом, разбудит своими лучами заснувшие за ночь растения, оно сделает их способными исполнять свою целительную функцию – исправлять воздух для животных; во мраке ночи эта деятельность совсем прекращается; днем же совершается с тем большим оживлением, чем светлее день и чем выгоднее расположено растение в отношении солнечных лучей. Затененные высокими зданиями или другими растениями, они не исправляют воздух, а, наоборот, выделяют вредный для дыхания животных воздух. К концу дня выработка очищенного воздуха ослабевает и при заходе солнца совершенно прекращается».
Первые точные исследования процесса дыхания у растений принадлежат Соссюру (1804). Он брал свежие листья и помещал их на ночь в сосуд, наполненный воздухом. При этом кислород воздуха поглощался и выделялся углекислый газ. Если на следующий день листья снова выставлялись на солнечный свет, то они выделяли почти такое же количество кислорода, какое поглотили ночью. Свои исследования Соссюр распространил и на незеленые части растений: стебли древесных растений, цветки, корни, плоды, и доказал, что дыхание наблюдается также в клетках этих органов. Он обнаружил, что при дыхании потеря в весе растения равна весу выделенного углерода.
Соссюр обратил внимание и на то, что молодые, растущие части растения, например новые побеги и распускающиеся цветки, дышат интенсивнее и потребляют кислорода больше, чем части растения, прекратившие рост.
Если, по Лавуазье, дыхание имеет сходство с процессом горения, то каким же образом органические вещества могут «гореть» при обычной температуре тела организма, да еще в водной среде, (ведь на 70 — 90% масса живых организмов состоит из воды)? Возникло предположение о том, что в живых клетках существуют механизмы, активирующие кислород. Швейцарский химик X. Ф. Шейнбайн, открывший озон, изучал причины быстрого потемнения пораненной поверхности растительных тканей, таких, как ткани яблок, картофеля, плодовых тел грибов. В 1845 г. он выступил со своей теорией окислительных процессов, согласно которой в живых клетках имеются соединения, способные легко окисляться в присутствии 02 и таким образом активировать молекулярный кислород. Если ткань прокипятить, то потемнения не происходит. Следовательно, потемнение тканей — каталитический окислительный процесс. Шейнбайн ошибочно полагал, что активация кислорода — это образование озона.
Исследования, начатые Шейнбайном, продолжил А. Н. Бах, который в 1897 г. разработал перекисную теорию биологического окисления, приложив ее к процессам дыхания. Несколько позже, в том же 1897 г., аналогичные взгляды высказал немецкий исследователь К. Энглер.
Суть перекисной теории биологического окисления Баха заключается в следующем. Молекулярный кислород имеет двойную связь и для того чтобы его активировать, необходимо эту двойную связь расщепить. Легко окисляющееся соединение А взаимодействует с кислородом и, разрывая двойную связь, образует пероксид А02 Таким образом, по мысли Баха, активация кислорода есть образование пероксида. В свою очередь пероксидное соединение, взаимодействуя с соединением В, окисляет его; затем эта реакция повторяется со вторым атомом кислорода и второй молекулой соединения В. Получается полностью восстановленное исходное соединение — акцептор кислорода А и полностью окисленное вещество В.
Много позднее, в 1955 г., две группы исследователей — О. Хаяиши с сотр. в Японии и Г. С. Мэзон с сотр. в США, используя современные методы, проанализировали возможность включения кислорода в органические соединения.
В настоящее время известно, что путь включения кислорода в органические соединения в соответствии с перекисной теорией биологического окисления Баха и Энглера не имеет отношения к дыханию, однако работы этих исследователей сыграли большую роль в изучении химизма дыхания, заложив основы современного понимания механизмов активации кислорода.
История современного учения о дыхании растений неразрывно связана с именем академика В.И. Палладина.
В годы первого петербургского периода работы Палладин исследовал ферментативную природу дыхательного процесса. Палладин показал, что и анаэробная, и аэробная фазы дыхания обеспечиваются специфическими ферментами, последовательно перерабатывающими продукты дыхания. Итоги работ этого периода изложены в монографии В.И. Палладина «Дыхание как сумма ферментативных процессов» (1907).
Одновременно с Палладиным проблемой дыхания занимались в целом ряде крупнейших научно-исследовательских институтов и лабораторий Западной Европы. Наибольшую популярность приобрели две новые школы – Виланда и Варбурга.
Т. Виланд развивал взгляды на роль дегидраз и водородных акцепторов, вполне аналогичные взглядам Палладина. Расхождение их теорий заключалось в том, что Виланд категорически отрицал какую бы то ни было роль оксидаз как специфических активаторов кислорода, считая молекулярный кислород способным самостоятельно отнимать водород от водородного акцептора. По мнению же Палладина, водородные акцепторы не могут самопроизвольно освобождаться от водорода, но требуют для этого участия оксидаз, которые поэтому являются обязательным фактором в реакции, выраженной во втором уравнении Палладина.
Противник Виланда, Варбург, считал, что молекулярный кислород не может вступить в организме в какой бы то ни было окислительный процесс, если в организме отсутствует система железоорганических соединений, типичным представителем которых он считал геминфермент. Варбург утверждал, что геминфермент активирует молекулярный кислород, т.е. как бы дает первый толчок к началу окислительных процессов, и без него никакой дыхательный процесс не может совершаться. Далее, по мнению Варбурга, окислительный импульс через промежуточные звенья (геминовые соединения) доходит до дыхательного субстрата и окисляет его. Резюмируя свои взгляды, Варбург утверждал, что дыхание осуществляется путем активации кислорода, а отнюдь не водорода. Но ведь Палладин как раз и говорил о той же необходимости активации молекулярного кислорода, защищая перед Виландом роль оксидаз в процессе дыхания.
Все различие в основных посылках Варбурга и Палладина заключается в том, что первый, работая по преимуществу с объектами животного происхождения, называл свой активатор молекулярного кислорода геминферментом, а Палладин, работавший с объектами растительного происхождения, сохранил за этим активатором ранее установившееся в науке название оксидазы. Но по существу оба говорили об одном и том же, протестуя против непримиримой позиции Виланда, отрицавшего необходимость энзиматической активации молекулярного кислорода.
Английский биохимик Д. Кейлин в 1925 г. доказал присутствие в клетках цитохромоксидазы, ускоряющей поглощение ими кислорода, и открыл другие цитохромы. Затем цитохромы были обнаружены у всех аэробов и было показано, что у этих организмов на завершающем этапе процесса дыхания осуществляется перенос на кислород электронов и протонов, в результате чего образуется Н20 (или Н202).
3.Каталитические системы дыхания
Окисление дыхательных субстратов в ходе дыхания осуществляется с участием ферментов. Ферменты как белковые катализаторы, помимо свойств, присущих неорганическим катализаторам, обладают рядом особенностей: высокой активностью, высокой специфичностью по отношению к субстратам и высокой лабильностью. Их пространственная организации зависящая от нее активность изменяются под действием внешних и внутренних факторов. Эти свойства обеспечивают возможность тонкой регуляции обмена веществ на уровне ферментов.
Типы окислительно-восстановительных реакций. Существуют четыре способа окисления, и все они связаны с отнятием электронов:
1) непосредственная отдача электронов, например:
![]()
2) Отнятие водорода:
![]()
3) присоединение кислорода:
![]()
4) образование промежуточного гидратированного соединения с последующим отнятием двух электронов и протонов:
![]()
Оксидоредуктазы.
Поскольку окисление одного вещества (донора электронов и протонов) сопряжено с восстановлением другого соединения (их акцептора), ферменты, катализирующие эти реакции, называют оксидоредуктазами. Все они относятся к I классу ферментов:

Донор (Д) отдает электроны и протоны, акцептор (А) принимает их, а энзим (Е) осуществляет реакцию переноса. Существуют три группы оксидоредуктаз:
а) анаэробные дегидрогеназы передают электроны различным промежуточным акцепторам, но не кислороду;
б) аэробные дегидрогеназы передают электроны различным акцепторам, в том числе кислороду;
в) оксидазы способны передавать электроны только кислороду.
Анаэробные дегидрогеназы. Это двухкомпонентные ферменты, коферментом которых может быть НАД+ (никотинамидадениндинуклеотид):

При окислении субстрата НАД+ превращается в восстановленную форму НАДH, а второй протон субстрата диссоциирует в среду (НАДH+ Н+). К анаэробным НАД-зависимым дегидрогеназам относятся такие ферменты, как алкогольдегидрогеназа, лактатдегидрогеназа, малатдегидрогеназа и др. Коферментом анаэробных дегидрогеназ может быть также НАДФ+ (никотинамидадениндинуклеотидфосфат), содержащий на одну фосфатную группировку больше, чем НАД + . НАДФ- зависимыми дегидрогеназами являются изоцитратдегидрогеназа, глюкозо-6-фосфатдегидрогеназа, 6-фосфоглюконатдегидрогеназа и др.
Субстратная специфичность фермента зависит от его белковой части. Многие НАД- и НАДФ-зависимые дегидрогеназы нуждаются в присутствии ионов двухвалентных металлов. Например, алкогольдегидрогеназа содержит ионы цинка.
Окисленные и восстановленные формы коферментов анаэробных дегидрогеназ могут взаимопревращаться в реакции, катализируемой ферментом НАД(Ф)-трансгидрогеназой:
НАДФH + НАД+ = НАДФ+ + НАДH
Анаэробные дегидрогеназы передают водород, т. е. электроны и протоны, различным промежуточным переносчикам и аэробным дегидрогеназам.
Аэробные дегидрогеназы. Это также двухкомнонентные ферменты, получившие название флавиновых (флавопротеины).
Помимо белков, в их состав входит прочно связанная с ними простетическая группа — рибофлавин (витамин В2).
Различают два кофермента этой группы: флавинмононуклеотид (ФМН), или желтый дыхательный фермент Варбурга, и флавинадениндинуклеотид (ФАД).
ФМН (рибофлавин-5-фосфат) содержит гетероциклическое азотистое основание — диметилизоаллоксазин, спирт рибит (производное рибозы) и фосфат:

В ФАД кроме ФМН имеется еще один нуклеотид — аденозинмонофосфата:

Активной группой в реакции присоединения и отдачи электронов и протонов в ФМН и ФАД служит изоаллоксазин. Взаимодействие с восстановленным переносчиком, например НАДH, происходит следующим образом:
Примером дегидрогеназы, в состав которой входит ФАД, является сукцинатдегидрогеназа. Доноры электронов для аэробных дегидрогеназ — анаэробные дегидрогеназы, а акцепторы — хиноны, цитохромы, кислород.
Цитохромная система. Среди оксидаз очень важную роль играют железосодержащие ферменты и переносчики, относящиеся к цитохромной системе. В нее входят цитохромы " и цитохромоксидаза. Включаясь в определенной последовательности в процесс переноса электронов, они передают их от флавопротеинов на молекулярный кислород.
Все компоненты цитохромной системы содержат железопорфириновую простетическую группу.
При переносе электронов цитохромами железо обратимо окисляется и восстанавливается, отдавая или приобретая электрон и изменяя таким образом свою валентность. В дыхательной цепи направление транспорта электронов определяется величиной окислительно-восстановительного потенциала цитохромов.
В этой системе передавать электроны непосредственно на кислород способна только цитохромоксидаза (цит. а + а3). Из всех известных оксидаз она имеет наибольшее сродство к кислороду. Ингибиторами цитохромоксидазы являются СО, цианид, азид. Б растительных митохондриях кроме цитохромоксидазы функционирует оксидаза, не подавляемая цианидом и названная альтернативной оксидазой. Например, в митохондриях початков ароидных активность цианидустойчивой оксидазы в 10 раз превышает активность цитохромоксидазы.
Пероксидаза и каталаза. К пероксидазам относят целую группу ферментов, использующих в качестве окислителя пероксид водорода: классическую пероксидазу, НАД-пероксидазу, НАДФ-пероксидазу, пероксидазу жирных кислот, глутатионпероксидазу, цитохромпероксидазу и др. Все они работают по следующей схеме, где А — субстраты:
![]()
В последние 2 — 3 десятилетия показана полифункциональность пероксидаз. Помимо пероксидазной, у них имеется оксидазная функция, т. е. способность переносить электроны в отсутствие пероксидного кислорода на молекулярный кислород. Пероксидаза может также функционировать как анаэробная дегидрогеназа, например НАДH-дегидрогеназа, передающая электроны от восстановленных пиридиновых нуклеотидов на разные акцепторы.
Пероксид водорода, помимо пероксидазы, расщепляется также каталазой, в результате чего образуется молекулярный кислород. В реакции участвуют две молекулы пероксида, одна из которых функционирует как донор, а другая — как акцептор электронов.
Простетической группой пероксидазы и каталазы служит гем, в состав которого входит атом железа.
Оксигеназы. Наряду с оксидазами, которые используют молекулярный кислород как акцептор электронов, в клетках широко представлены оксигеназы, активирующие кислород, в результате чего он может присоединяться к органическим соединениям. Ферменты, внедряющие в субстрат два атома кислорода, называют диоксигеназами, а присоединяющие один атом кислорода — монооксигеназами или гидроксилазами. В качестве доноров электронов оксигеназы используют НАД(Ф)H, ФАДH2 и др.
Оксигеназы присутствуют во всех типах клеток. Они участвуют в гидроксилировании многих эндогенных соединений в частности аминокислот, фенолов, стеринов и др., а также в детоксикации чужеродных токсических веществ (ксенобиотиков).
4.Основные пути диссимиляции углерода
Существуют два основных пути окисления углеводов: 1) дихотомический (гликолитический) и 2) апотомический (пентозофосфатный). Белки, жиры и органические кислоты окисляются в глиоксилатном цикле.
Относительная роль этих путей дыхания может меняться в зависимости от типа растений, возраста, фазы развития, а также в зависимости от условий внешней среды. Процесс дыхания растений осуществляется во всех внешних условиях, в которых возможна жизнь. Растительный организм не имеет приспособлений к регуляции температуры, поэтому процесс дыхания осуществляется при температуре от – 50 до +50°С. Нет приспособлений у растений и к поддержанию равномерного распределения кислорода по всем тканям. Именно необходимость осуществления процесса дыхания в разнообразных условиях привела к выработке в процессе эволюции разнообразных путей дыхательного обмена и к ещё большему разнообразию ферментативных систем, осуществляющих отдельные этапы дыхания. При этом важно отметить взаимосвязь всех процессов обмена в организме. Изменение пути дыхательного обмена приводит к глубоким изменениям во всем метаболизме растительных организмов.
4.1 Дихотомический путь
Это основной путь распада органических веществ для всех живых организмов. Выделяют 2 этапа дихотомического пути: гликолиз и цикл Кребса.

Рис. 1 Основные этапы дыхания
4.1.1Гликолиз. Механизмы регуляции цикла. Энергетическая эффективность процесса, значение. Связь с другими процессами
Гликолиз — процесс анаэробного распада глюкозы, идущий Гликолиз с освобождением энергии, конечным продуктом которого является пировиноградная кислота. Гликолиз — общий начальный этап аэробного дыхания и всех видов брожения. Реакции гликолиза протекают в растворимой части цитоплазмы (цитозоле) и в хлоропластах. В цитозоле гликолитические ферменты, по-видимому, организованы в мультиэнзимные комплексы с участием актиновых филаментов цитоскелета, с которыми гликолитические ферменты обратимо связываются с разной степенью прочности. Такое связывание обеспечивает векторность процесса гликолиза.
Английский биохимик А. Гарден и ученик К. А. Тимирязева Л. А. Иванов в 1905 г. независимо показали, что в процессе спиртового брожения наблюдается связывание неорганического фосфата и превращение его в органическую форму. Гарден установил, что глюкоза подвергается анаэробному распаду только после ее фосфорилирования. Полностью весь процесс гликолиза расшифровали немецкие биохимики Г. Эмбден, О. Ф. Мейергоф и советский биохимик Я. О. Парнас, с именами которых связывают название этого катаболического
Цепь реакций, составляющих суть гликолиза, можно разбить на три этапа:
I. Подготовительный этап — фосфорилирование гексозы и ее расщепление на две фосфотриозы.
II. Первое субстратное фосфорилирование, которое начинается с 3-фосфоглицеринового альдегида и кончается 3-фосфоглицериновой кислотой. Окисление альдегида до кислоты связано с освобождением энергии. В этом процессе на каждую фосфотриозу синтезируется одна молекула АТФ.
III. Второе субстратное фосфорилирование, при котором 3-фосфоглицериновая кислота за счет внутримолекулярного окисления отдает фосфат с образованием АТФ.
Поскольку глюкоза стабильное соединение, на ее активацию необходима затрата энергии, что осуществляется в процессе образования фосфорных эфиров глюкозы в ряде подготовительных реакций. Глюкоза (в пиранозной форме) фосфорилируется АТФ с участием гексокиназы (1), превращаясь в глюкозо-6-фосфат, который изомеризуется в фруктозо-6-фосфат с помощью глюкозофосфатизомеразы (2).
Этот - переход необходим для образования более лабильной фуранозной формы молекулы гексозы. Фруктозо-6-фосфат фосфорилируется вторично фосфофрукгокиназой с использованием еще одной молекулы АТФ (3).
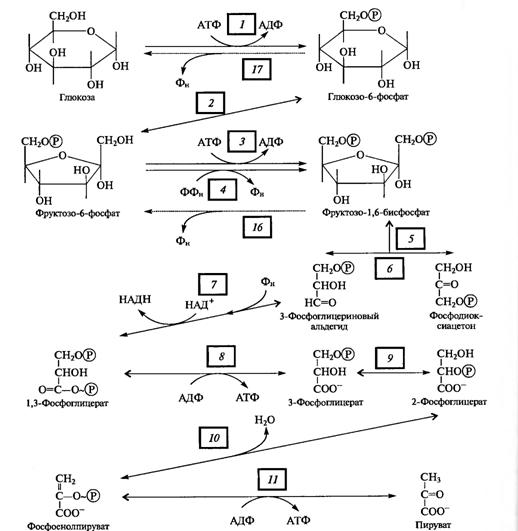
Рис. 2. Реакции гликолиза
Фруктозо-1,6-дифосфат — лабильная фуранозная форма с симметрично расположенными фосфатными группами. Обе эти группы несут отрицательный заряд, отталкиваясь друг от друга электростатически. Такая структура легко расщепляется альдолазой на две фосфотриозы. Следовательно, смысл подготовительного этапа состоит в активации молекулы гексозы за счет двойного фосфорилирования и перевода в фуранозную форму с последующим распадом на 3-фосфоглицериновый альдегид (3-ФГА) и фосфодиоксиацетон (ФДА) (5), причем бывший 6-й атом углерода в молекуле глюкозы и фруктозы (фосфорилированный) становится 3-м в 3-ФГК, а 1-й атом углерода фруктозо-1,6-дифосфата остается 1-м углеродом (фосфорилированным) в ФДА. 3-ФГА и ФДА легко превращаются друг в друга с участием триозофосфатизомеразы (6). Из-за расщепления молекулы гексозы на две триозы гликолиз иногда называютдихотомическим путем окисления глюкозы.
С 3-ФГА начинается II этап гликолиза — первое субстратное фосфорилирование. Фермент дегидрогеназа фосфоглицеринового альдегида (НАД-зависимый SH-фермент) (7) образует с 3-ФГА фермент-субстратный комплекс, в котором происходит окисление субстрата и передача электронов и протонов на НАД+. В ходе окисления фосфоглицеринового альдегида до фосфоглицериновой кислоты в фермент-субстратном комплексе возникает меркаптанная высокоэнергетическая связь (т. е. связь с очень высокой свободной энергией гидролиза). Далее осуществляется фосфоролиз этой связи, в результате чего SH-фермент отщепляется от субстрата, а к остатку карбоксильной группы субстрата присоединяется неорганический фосфат, причем ацилфосфатная связь сохраняет значительный запас энергии, освободившейся в результате окисления 3-ФГА. Высокоэнергетическая фосфатная группа с помощью фосфоглицераткиназы передается на АДФ и образуется АТФ (8). Так как в данном случае высокоэнергетическая ковалентная связь фосфата формируется прямо на окисляемом субстрате, такой процесс получил названиесубстратного фосфорилирования. Таким образом, в результате II этапа гликолиза образуются АТФ и восстановленный НАДH.
Последний этап гликолиза — второе субстратное фосфорилирование. З-Фосфоглицериновая кислота с помощью фосфоглицератмутазы превращается в 2-фосфоглицериновую кислоту (9). Далее фермент енолаза катализирует отщепление молекулы воды от 2-фосфоглицериновой кислоты (10). Эта реакция сопровождается перераспределением энергии в молекуле, в результате чего образуется фосфоенолпируват — соединение, содержащее высокоэнергетическую фосфатную связь. Таким образом, в этом случае высокоэнергетическая фосфатная связь формируется на основе того фосфата, который имелся в самом субстрате. Этот фосфат при участии пируваткиназы (11) передается на АДФ и образуется АТФ, а енолпируват самопроизвольно переходит в более стабильную форму — пируват — конечный продукт гликолиза.
Энергетический выход гликолиза. При окислении одной молекулы глюкозы образуются две молекулы пировиноградной кислоты. При этом за счет первого и второго субстратного фосфорилирования образуются четыре молекулы АТФ. Однако две молекулы АТФ тратятся на фосфорилирование гексозы на I этапе гликолиза. Таким образом, чистый выход гликолитического субстратного фосфорилирования составляет две молекулы АТФ.
Кроме того, на II этапе гликолиза на каждую из двух молекул фосфотриоз восстанавливается по одной молекуле НАДH. Окисление одной молекулы НАДH в электронтранспортной цепи митохондрий в присутствии 02 сопряжено с синтезом трех молекул АТФ, а в расчете на две триозы (т. е. на одну молекулу глюкозы) - шесть молекул АТФ. Таким образом, всего в процессе гликолиза (при условии последующего окисления НАДН) образуются восемь молекул АТФ. Поскольку свободная энергия гидролиза одной молекулы АТФ во внутриклеточных условиях составляет около 41,868 кДж/моль (10 ккал), восемь молекул АТФ дают 335 кДж/моль, или 80 ккал. Таков полный энергетический выход гликолиза в аэробных условиях.
Обращение гликолиза. Возможность обращения гликолиза определяется обратимостью действия большинства ферментов, катализирующих его реакции. Однако реакции фосфорилирования глюкозы и фруктозы, а также реакция образования пировиноградной кислоты из фосфоенолпирувата, осуществляемые с помощью киназ, необратимы. На этих участках процесс обращения идет благодаря использованию обходных путей. Там, где функционируют гексокиназа и фруктокиназа, происходит дефосфорилирование — отщепление фосфатных групп фосфатазами.
Превращение пирувата в фосфоенолпируват также не может осуществиться путем прямого обращения пируваткиназной реакции вследствие большого перепада энергии. Первая реакция обращения гликолиза на этом участке катализируется митохондриальной пируваткарбоксилазой в присутствии АТФ и ацетил-СоА (последний выполняет функции активатора). Образующаяся щавелевоуксусная кислота (ЩУК), или оксалоацетат, восстанавливается затем в митохондриях до малата при участии НАД-зависимой малатдегидрогеназы (МДГ). Затем малат транспортируется из митохондрий в цитоплазму, где окисляется НАД-зависимой цитоплазматической малатдегидрогеназой снова до ЩУК. Далее под действием ФЕП-карбоксикиназы из оксалоацетата образуется фосфоенолпируват. Фосфорилирование в этой реакции осуществляется за счет АТФ.
Значение гликолиза в клетке. В аэробных условиях гликолиз выполняет ряд функций: 1) осуществляет связь между дыхательными субстратами и циклом Кребса; 2) поставляетнанужды клетки две молекулы АТФ и две молекулы НАДHпри окислении каждой молекулы глюкозы (в условиях аноксии гликолиз, по-видимому, служит основным источником АТФ в клетке); 3) производит интермедиа, необходимые для синтетических процессов в клетке (например, фосфоенолпируват, необходимый для образования фенольных соединений и лигнина); 4) в хлоропластах гликолитические реакции обеспечивают прямой путь для синтеза АТФ, независимый от поставок НАДФH; кроме того, через гликолиз в хлоропластах запасенный крахмал метаболизируется в триозы, которые затем экспортируются из хлоропласта.
Регуляция гликолиза.
Интенсивность гликолиза контролируется в нескольких участках. Вовлечение глюкозы в процесс гликолиза регулируется на уровне Фермента гексокиназы по типу обратной связи: избыток продукта реакции (глюкозо-6-фосфата) аллостерически подавляет деятельность фермента.
Второй участок регуляции скорости гликолиза находится на уровне фосфофруктокиназы. Фермент аллостерически ингибируется высокой концентрацией АТФ и активируется неорганическим фосфатом и АДФ. Ингибирование АТФ предотвращает развитие реакции в обратном направлении при высокой концентрации фруктозо-6-фосфата. Кроме того, фермент подавляется продуктом цикла Кребса — цитратом и через положительную обратную связь активируется собственным продуктом — фруктозо-1,6-дифосфатом (самоусиление).
Высокие концентрации АТФ подавляют активность пируваткиназы, снижая сродство фермента к фосфоенолпирувату. Пируваткиназа подавляется также ацетил-СоА.
Наконец, пируватдегидрогеназный комплекс, участвующий в образовании ацетил-СоА из пирувата, ингибируется высокими концентрациями АТФ, а также НАДH и собственным продуктом — ацетил-СоА.
4.1.2 Цикл Кребса. Механизмы регуляции цикла. Энергетическая эффективность процесса, значение
В анаэробных условиях пировиноградная кислота (пируват) подвергается дальнейшим превращениям в ходе спиртового, молочнокислого и других видов брожений, при этом НАДH используется для восстановления конечных продуктов брожения, регенерируя в окисленную форму. Последнее обстоятельство поддерживает процесс гликолиза, для которого необходим окисленный НАД + . В присутствии достаточного количества кислорода пируват полностью окисляется до С02 и Н20 в дыхательном цикле, получившем названиецикла Кребса, цикла ди- или трикарбоновых кислот. Все участки этого процесса локализованы в мАТФиксе или во внутренней мембране митохондрий.
Последовательность реакций в цикле Кребса. Участие органических кислот в дыхании давно привлекало внимание исследователей. Еще в 1910 г. шведский химик Т. Тунберг показал, что в животных тканях содержатся ферменты, способные отнимать водород от некоторых органических кислот (янтарной, яблочной, лимонной). В 1935 г. А. Сент-Дьердьи в Венгрии установил, что добавление к измельченной мышечной ткани небольших количеств янтарной, фумаровой, яблочной или щавелевоуксуснсй кислот резко активирует поглощение тканью кислорода.
Учитывая данные Тунберга и Сент-Дьердьи и исходя из собственных экспериментов по изучению взаимопревращения различных органических кислот и их влияния на дыхание летательной мышцы голубя, английский биохимик Г. А. Кребс в 1937 г. предложил схему последовательности окисления ди- и трикарбоновых кислот до С02 через«цикл лимонной кислоты» да счет отнятия водорода. Этот цикл и был назван его именем.
Непосредственно в цикле окисляется не сам пируват, а его производное — ацетил-СоА. Таким образом, первым этапом на пути окислительного расщепления ПВК является процесс образования активного ацетила в ходе окислительного декарбоксилирования. Окислительное декарбоксилирование пирувата осуществляется при участии пируватдегидрогеназного мультиферментного комплекса. В состав его входят три фермента и пять коферментов. Коферментами служат тиаминпирофосфат (ТПФ) — фосфорилированное производное витамина Вь липоевая кислота, коэнзим A, ФАД и НАД+. Пируват взаимодействует с ТПФ (декарбоксилазой), при этом отщепляется С02 и образуется гидроксиэтильное производное ТПФ (рис. 3). Последнее вступает в реакцию с окисленной формой липоевой кислоты. Дисульфидная связь липоевой кислоты разрывается и происходит окислительно-восстановительная реакция: гидроксиэтильная группа, присоединенная к одному атому серы, окисляется в ацетильную (при этом возникает высокоэнергетическая тиоэфирная связь), а другой атом серы липоевой кислоты восстанавливается. Образовавшаяся ацетиллипоевая кислота взаимодействует с коэнзимом А, возникают ацетил- СоА и восстановленная форма липоевой кислоты. Водород липоевой кислоты переносится затем на ФАД и далее на НАД + . В результате окислительного декарбоксилирования пирувата образуются ацетил-СоА, С02 и НАДH.

Рис. 3. Окислительное декарбоксилирование ПВК
Дальнейшее окисление ацетил-СоА осуществляется в ходе циклического процесса.
Цикл Кребса начинается с взаимодействия ацетил-СоА с енольной формой щавелевоуксусной кислоты. В этой реакции под действием фермента цитратсинтазы образуется лимонная кислота (2). Следующий этап цикла включает две реакции и катализируется ферментом аконитазой, или аконитатгидратазой (3). В первой реакции в результате дегидратации лимонной кислоты образуется цис-аконитовая. Во второй реакции аконитат гидратируется и синтезируется изолимонная кислота. Изолимонная кислота под действием НАД- или НАДФ-зависимой изоцитратдегидрогеназы (4) окисляется в нестойкое соединение — щавелевоянтарную кислоту, которая тут же декарбоксилируется с образованием α-кетоглутаровой кислоты (α-оксоглутаровой кислоты).
α-Кетоглутарат, подобно пирувату, подвергается реакции окислительного декарбоксилирования. α-Кетоглутаратдегидрогеназный мультиэнзимный комплекс (5) сходен с рассмотренным выше пируватдегидрогеназным комплексом. В ходе реакции окислительного декарбоксилирования α-кетоглутарата выделяется С02, образуются НАДH и сукцинил-СоА.

Рис. 4. Цикл Кребса
Подобно ацетил-СоА, сукцинил-СоА является высокоэнергетическим тиоэфиром. Однако если в случае с ацетил-СоА энергия тиоэфирной связи расходуется на синтез лимонной кислоты, энергия сукцинил-CoA может трансформироватся в образование фосфатной связи АТФ. При участии сукцинил- СоА-синтетазы (6) из сукцинил-СоА, АДФ и Н3Р04 образуются янтарная кислота (сукцинат), АТФ, регенерирует молекула СоА. АТФ образуется в результате субстратного фосфорилирования.
На следующем этапе янтарная кислота