Химические свойства серной кислоты
Химические свойства серной кислоты такие:
1. Взаимодействие с металлами:
· разбавленная кислота растворяет только те металлы, которые стоят левее водорода в ряду напряжений, например H2+1SO4+ Zn0 = H2O + Zn+2SO4;
· окислительные свойства серной кислоты велики. При взаимодействии с различными металлами (кроме Pt, Au) она может восстанавливаться до H2S-2 , S+4O2 или S0, например:
· 2H2+6SO4 + 2Ag0 = S+4O2 + Ag2+1SO4 + 2H2O;
· 5H2+6SO4 +8Na0 = H2S-2 + 4Na2+1SO4 + 4H2O;
2. Концентрированная кислота H2S+6O4также реагирует (при нагревании) с некоторыми неметаллами, превращаясь при этом в соединения серы с более низкой степенью окисления, например:
· 2H2S+6O4 + С0 = 2S+4O2 + C+4O2 + 2H2O;
· 2H2S+6O4 + S0 = 3S+4O2 + 2H2O;
· 5H2S+6O4 + 2P0 = 2H3P+5O4 + 5S+4O2 + 2H2O;
3. С основными оксидами:
· H2SO4 + CuO = CuSO4 + H2O;
4. С гидроксидами:
· Cu(OH)2 + H2SO4 = CuSO4 + 2H2O;
· 2NaOH + H2SO4 = Na2SO4 + 2H2O;
5. Взаимодействие с солями при обменных реакциях:
· H2SO4 + BaCl2 = 2HCl + BaSO4;
Образование BaSO4 (белого осадка, нерастворимого в кислотах) используется для определения этой кислоты и растворимых сульфатов.
Моногидрат - это ионизирующий растворитель, имеющий кислотный характер. В нём очень хорошо растворять сульфаты многих металлов, например:
· 2H2SO4 + HNO3 = NO2+ + H3O+ + 2HSO4-;
· HClO4 + H2SO4 = ClO4- + H3SO4+.
Концентрированная кислота - это довольно сильный окислитель, особенно при нагревании, например 2H2SO4 + Cu = SO2 + CuSO4 + H2O.
Действуя как окислитель, серная кислота, как правило, восстанавливается до SO2. Но она может быть восстановлена и до S и даже до H2S, например H2S + H2SO4 = SO2 + 2H2O + S.
Моногидрат почти не может проводить электрический ток. И, наоборот, водные растворы кислоты - это хорошие проводники. Серная кислота сильно поглощает влагу, поэтому ее используют для осушки разных газов. Как осушитель, серная кислота действует до тех пор, пока над её раствором давление водяного пара меньше, чем его давление в газе, который осушают.
Если закипятить разбавленный раствор серной кислоты, то из него уберется вода, при этом температура кипения будет повышаться до 337°С, например, когда начинают перегонять серную кислоту в концентрации 98,3%. И наоборот, из растворов, которые более концентрированные, испаряется лишний серный ангидрид. Пар кипящей при температуре 337°С кислоты частично разложен на SO3 и H2O, которые при охлаждении опять будут соединены. Высокая температура кипения этой кислоты подходит для использования её в выделении легколетучих кислот из их солей при нагревании.
10. Теория электролитической диссоциации
Обобщим сведения об электролитической диссоциации в виде основных положений ныне общепризнанной теории. Они заключается в следующем.
В результате такого взаимодействия образуются гидратированные, то есть связанные с молекулами воды, ионы.
Следовательно, по наличию водной оболочки ионы делятся на гидратированные (в растворах и кристаллогидратах) и не-гидратированные (в безводных солях). Свойства гидратиронянных и негндратировашшх ионов отличаются, как вы смогли уже убедиться на примере ионов меди.
При растворении в воде электролиты диссоциируют (расспадаются) на положительные и отрицательные ионы.Свойства ионов совершенно не похожи на свойства атомов, которые их образовали.
Ионы— это одна из форм существования химического элемента.
Например, атомы металла натрия энергично взаимодействуют с водой, образуя при этом щелочь и водород Н, в то время как ионы натрия таких продуктов не образуют. Хлор имеет желто-зеленый цвет и резкий запих, ядовит, а ионы хлора — бесцветны, неядовиты, лишены запаха.
Никому не придет в голову использовать в пищу металлический натрий и газообразный хлор, в то время как без хлорида натрия, состоящего из ионов натрия и хлора, невозможно приготовление пищи.
3. Под действием электрического тока положительно заряженные ионы движутся к отрицательному полюсу источника тока — катоду, и поэтому называются катионами, а отрицательно заряженные ионы движутся к положительному полюсу источника тока — аноду, и поэтому называются анионами. Следовательно, существует еще одна классификация ионов — по знаку их заряда. Сумма зарядов катионов равна сумме зарядов анионов, вследствие чего растворы электролитов остаются электронейтральными.
Ионы — это положительно или отрицательно заряженные частицы, в которые превращаются атомы или группы атомов одного или нескольких химических элементов в результате отдачи или присоединения электронов.
Само слово «ион» в переводе с греческого означает "странствующий". В растворах ноны беспорядочно передвигаются («странствуют») в различных направлениях.По составу воны делятся на простые и сложные.
2. Причиной диссоциации электролита вводных растворах является его гидратация, то есть взаимодействие электролита с молекулами воды и разрыв химической связи в нем.
4. Электролитическая диссоциация — процесс обратимый для слабых электролитов.
Наряду с процессом диссоциации (распад электролита на ионы) протекает и обратный процесс — ассоциация (соединение ионов). Поэтому в уравнениях электролитической диссоциации вместо знака равенства ставят знак обратимости, например:
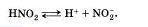
5. Не все электролиты в одинаковой мере диссоциируют на ионы.
Степень диссоциации зависит от природы электролита и его концентрации. По степени диссоциации электролиты делят на сильные и слабые.
6. Химические свойства растворов электролитов определяются свойствами тех ионов, которые они образуют при диссоциации.
По характеру образующихся ионов различают три типа электролитов: кислоты, основания и соли.
7. Кислотами называют электролиты, которые при диссоциации образуют катионы водорода и анионы кислотного остатка:

Следует учитывать, что диссоциация электролитов по второй ступени происходит намного слабее и равновесие сдвинуто влево. Диссоциация по третьей ступени при обычных условиях не происходят.
Все кислоты объединяет то. что они при диссоциации обязательно образуют катионы водорода. Поэтому логично предположить, что общие характерные свойства кислот — кислый вкус, изменение окраски индикаторов и др. — обусловлены именно катионами водорода.
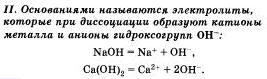
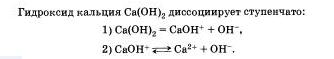
Очевидно, что свойства солей определяются как катионами металла, так и анионами кислотного остатка. Так. соли аммония имеют как общие свойства, обусловленные ионами, так и специфические, обусловленные различными анионами. Аналогично, общие свойства сульфатов — солей серной кислоты — определяются ионами SO 2- 4+ , а различные — разными катионами. В отличие от многоосновных кислот и оснований, содержащих несколько гидроксид-нонов, такие соли, как К2S04, АlCl3 и т.д. диссоциируют сразу полностью, а не ступенчато.
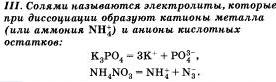
11. КЕКУЛЕ (Kekule), Фридрих Август
7 сентября 1829 г. – 13 июля 1896 г.
Немецкий химик Фридрих Август Кекуле фон Штрадониц родился в Дармштадте в семье чиновника. В юности Кекуле собирался стать архитектором. Он начал изучать архитектуру в Гисенском университете, но, прослушав курс лекций Ю. Либиха в дармштадтском Высшем техническом училище, заинтересовался химией. В 1849 г. Кекуле начал изучение химии у Либиха; после окончания университета в 1852 г. Кекуле уехал в Париж, где занимался химией у Ж. Дюма,А. Вюрца и Ш. Жерара. По возвращении в Германию Кекуле основал небольшую химическую лабораторию в Гейдельберге. Был приват-доцентом в Гейдельбергском (1856–1858) и профессором в Гентском (1858–1865) университетах. С 1865 г. до конца жизни занимал должность профессора Боннского университета, в котором некоторое время исполнял также обязанности ректора.
Экспериментальные работы Кекуле относятся к органической химии. В 1854 г. он получил тиоуксусную, а в 1856 г. – гликолевую кислоту. В 1872 г. совместно с нидерландским химиком А. Франшимоном (1844–1919) Кекуле синтезировал трифенилметан и антрахинон. С целью проверки гипотезы о равноценности всех атомов водорода в бензоле он получил его галоген-, нитро-, амино- и карбоксипроизводные; занимался также исследованиями ненасыщенных кислот и синтетических красителей. Однако основные работы Кекуле были посвящены теоретической химии; главной его заслугой стало создание теории валентности.
Мысль о том, что атом элемента обладает способностью к «насыщению», была высказана в 1853 г. Э. Франклендом при рассмотрении конституции металлорганических соединений. Развивая эту идею, в 1854 г. Кекуле впервые высказал идею о «двухосновности», или «двухатомности» (позднее он стал использовать термин «валентность») серы и кислорода, а в 1857 г. разделил все элементы на одно-, двух- и трехосновные; углерод Кекуле (одновременно с немецким химиком Г. Кольбе) определил как четырёхатомный элемент. В 1858 г. Кекуле (одновременно с шотландским химиком А. Купером) указал на способность атомов углерода при насыщении своих «единиц сродства» образовывать цепи. Это механическое учение о соединении атомов в цепи с образованием молекул легло в основу теории химического строения.
В 1865 г. Кекуле высказал предположение, что молекула бензола имеет форму правильного шестиугольника, образованного шестью углеродными атомами, с которыми связаны шесть атомов водорода. Объединив представление об образовании цепей с учением о существовании кратных связей, он пришел к идее чередования в бензольном кольце простых и двойных связей (сходные структурные формулы предложил незадолго до этогоИ. Лошмидт). Несмотря на то, что эта теория сразу столкнулась с возражениями, она довольно быстро привилась в науке и практике. Концепция Кекуле открыла путь к установлению структуры многих циклических (ароматических) соединений. Для объяснения неспособности бензола присоединять галогенводороды Кекуле в 1872 г. выдвинул осцилляционную гипотезу, согласно которой в бензоле простые и двойные связи постоянно меняются местами. В 1867 г. Кекуле опубликовал работу о пространственном расположении атомов в молекуле, где указывал, что связи углеродного атома могут не находиться в одной плоскости.
Кекуле несколько лет был президентом Немецкого химического общества. Он являлся одним из организаторов Международного конгресса химиков в Карлсруэ (1860). Весьма плодотворной была педагогическая деятельность Кекуле. Он автор получившего широкую известность «Учебника органической химии» (1859–1861). Целый ряд учеников Кекуле стали выдающимися химиками; среди них можно особо отметить Л. Мейера, Я. Вант-Гоффа, А. Байера и Э. Фишера.
12. БУТЛЕРОВ, Александр Михайлович
3(15) сентября 1828 г. – 5(17) августа 1886 г.
Русский химик Александр Михайлович Бутлеров родился в Чистополе Казанской губернии в семье помещика, офицера в отставке. Рано лишившись матери, Бутлеров воспитывался в одном из частных пансионов в Казани, затем учился в Казанской гимназии. В шестнадцатилетнем возрасте он поступил на физико-математическое отделение Казанского университета, который в то время был центром естественнонаучных исследований в России. В первые годы студенчества Бутлеров увлекался ботаникой и зоологией, но затем под влиянием лекций К. К. Клауса и Н. Н. Зинина заинтересовался химией и решил посвятить себя этой науке. В 1849 г. Бутлеров окончил университет и по представлению Клауса был оставлен на кафедре в качестве преподавателя. В 1851 г. он защитил магистерскую диссертацию «Об окислении органических соединений», а в 1854 г. – докторскую диссертацию «Об эфирных маслах». В 1854 г. Бутлеров стал экстраординарным, а в 1857 г. – ординарным профессором химии Казанского университета.
Во время заграничной поездки в 1857-1858 гг. Бутлеров познакомился со многими ведущими химиками Европы, участвовал в заседаниях только что организованного Парижского химического общества. В лаборатории Ш. А. Вюрца Бутлеров начал цикл экспериментальных исследований, послуживший основой теории химического строения. Её главные положения он сформулировал в докладе «О химическом строении вещества», прочитанном на Съезде немецких естествоиспытателей и врачей в Шпейере (сентябрь 1861 г.). Основы этой теории сформулированы таким образом: 1) «Полагая, что каждому химическому атому свойственно лишь определённое и ограниченное количество химической силы (сродства), с которой он принимает участие в образовании тела, я назвал бы химическим строением эту химическую связь, или способ взаимного соединения атомов в сложном теле»; 2) «... химическая натура сложной частицы определяется натурой элементарных составных частей, количеством их и химическим строением».
С этими постулатами прямо или косвенно связаны и все остальные положения классической теории химического строения. Бутлеров намечает путь для определения химического строения и формулирует правила, которыми можно при этом руководствоваться. Предпочтение он отдаёт синтетическим реакциям, проводимым в условиях, когда радикалы, в них участвующие, сохраняют своё химическое строение. Оставляя открытым вопрос о предпочтительном виде формул химического строения, Бутлеров высказывался об их смысле: «... когда сделаются известными общие законы зависимости химических свойств тел от их химического строения, то подобная формула будет выражением всех этих свойств». При этом Бутлеров был убеждён, что структурные формулы не могут быть просто условным изображением молекул, а должны отражать их реальное строение. Он подчёркивал, что каждая молекула имеет вполне определённую структуру и не может совмещать несколько таких структур.
Большое значение для становления теории химического строения имело её экспериментальное подтверждение в работах как самого Бутлерова, так и его школы. Бутлеров предвидел, а затем и доказал существование позиционной и скелетной изомерии. Получив третичный бутиловый спирт, он сумел расшифровать его строение и доказал (совместно с учениками) наличие у него изомеров. В 1864 г. Бутлеров предсказал существование двух бутанов и трёх пентанов, а позднее и изобутилена. Им было высказано также предположение о существовании четырех валериановых кислот; строение первых трёх было определено в 1871 г. Э. Эрленмейером, а четвёртая получена самим Бутлеровым в 1872 г. Чтобы провести идеи теории химического строения через всю органическую химию, Бутлеров издал в 1864-1866 гг. в Казани книгу «Введение к полному изучению органической химии», 2-е изд. которой вышло уже в 1867-1868 гг. на немецком языке.
В 1868 г. по представлению Д. И. Менделеева Бутлеров был избран ординарным профессором Петербургского университета, где и работал до конца жизни. В 1870 г. он стал экстраординарным, а в 1874 г. – ординарным академиком Петербургской академии наук. С 1878 по 1882 г. был Президентом и председателем Отделения химии Русского физико-химического общества.
Преподавательская деятельность Бутлерова длилась 35 лет и проходила в трех высших учебных заведениях: Казанском, Петербургском университетах и на Высших женских курсах (он принимал участие в их организации в 1878 г.). Под руководством Бутлерова работало множество его учеников, среди которых можно назвать В. В. Марковникова,Ф. М. Флавицкого, А. М. Зайцева (в Казани), А. Е. Фаворского, И. Л. Кондакова (в Петербурге). Бутлеров стал основателем знаменитой казанской («бутлеровской») школы химиков-органиков. Бутлеров прочитал также множество популярных лекций, главным образом на химико-технические темы.
Кроме химии, Бутлеров много внимания уделял практическим вопросам сельского хозяйства, садоводству, пчеловодству, а позднее также и разведению чая на Кавказе. С конца 1860-х гг. Бутлеров активно интересовался спиритизмом и медиумизмом, которым посвятил несколько статей; это увлечение Бутлерова и его попытки дать спиритизму научное обоснование стали причиной его полемики с Менделеевым. Умер Бутлеров в дер. Бутлеровка Казанской губернии, не дожив до окончательного признания своей теории. Два наиболее значительных русских химика – Д. И. Менделеев и Н. А. Меншуткин – лишь спустя десять лет после смерти Бутлерова признали справедливость теории химического строения.
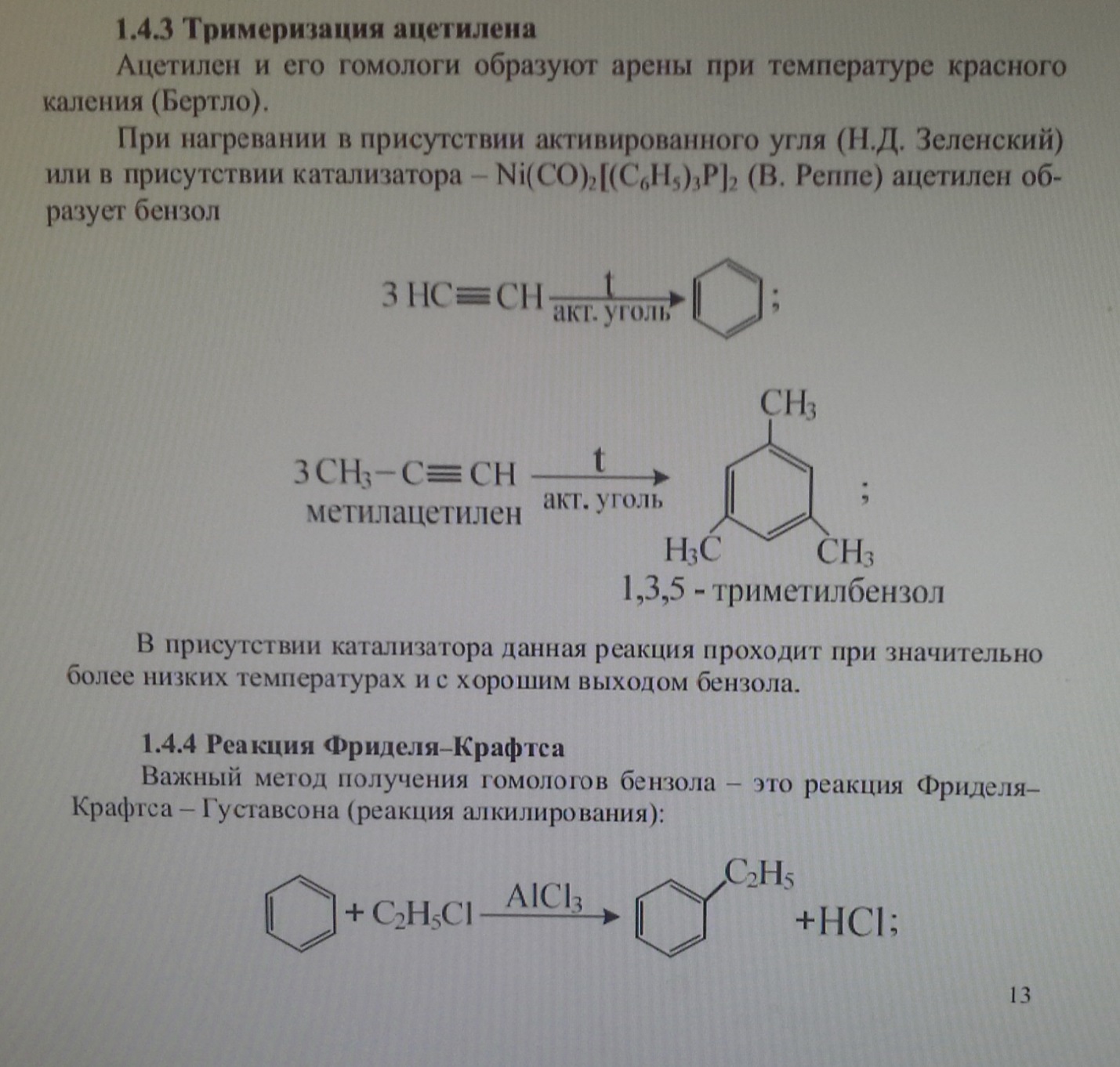
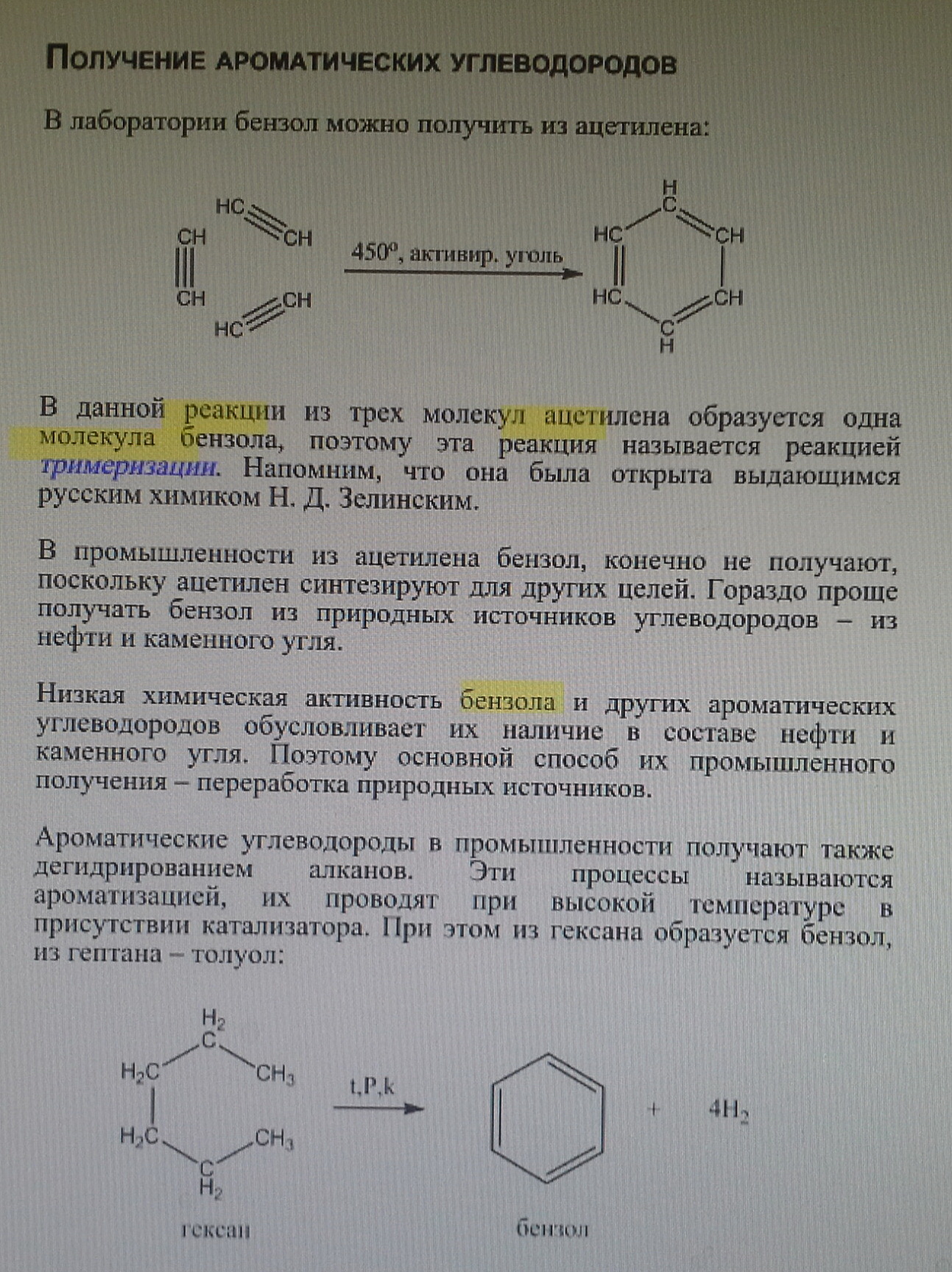
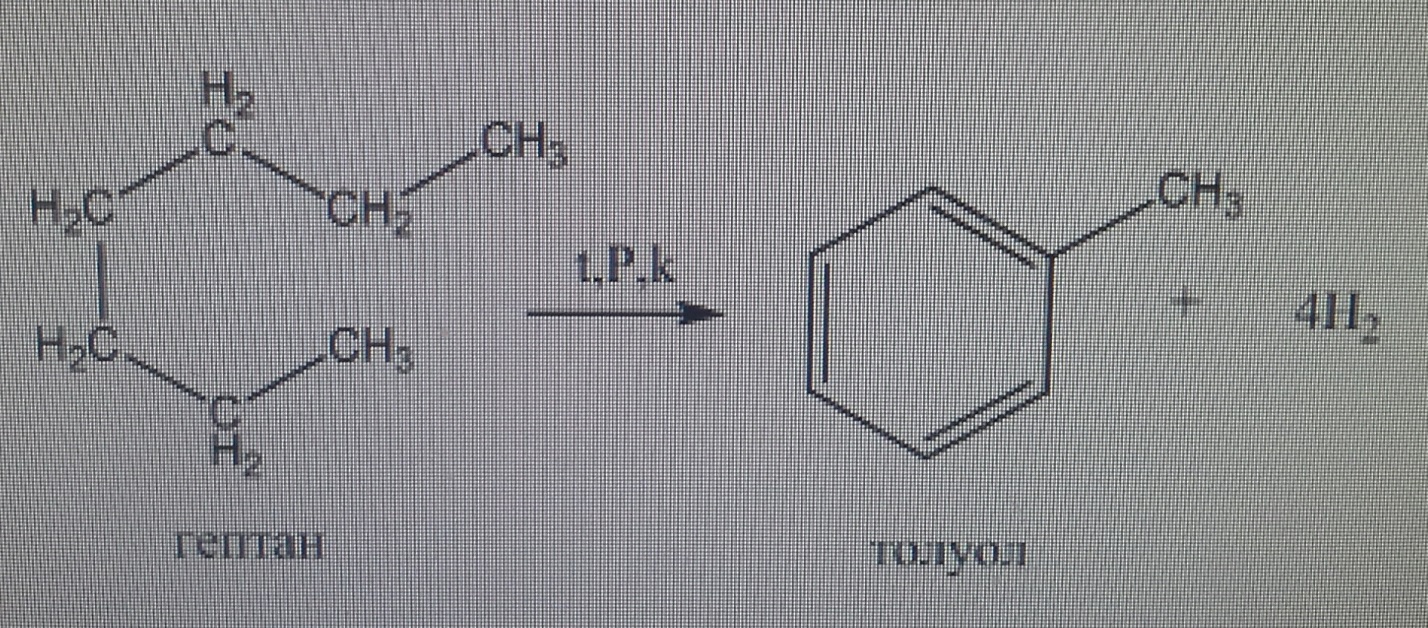
13. Реакция тримеризации бензола