Модель гармонического осциллятора
Для наглядности будем рассматривать простейший случай – колебание двухатомной молекулы, для определенности выберем молекулу хлористого водорода HCl. Решение любой задачи подразумевает выбор некоторой модели, по возможности более простой, но отражающей основные свойства реальной системы. Определимся с моделью двухатомной молекулы, объясняющей инфракрасные колебательные спектры поглощения.
В начале будем исходить из максимально упрощенной модели. Очевидно, таковой является система из двух материальных точек с массами, равными массам ядер молекулы, и противоположными зарядами. В случае молекул, состоящих из одинаковых атомов, например H2, материальные точки нашей модели не имеют заряда. В случае же молекулы HCl, точка, соответствующая водороду, имеет отрицательный заряд, а точка, соответствующая хлору – положительный. В данном приближении дипольный момент d молекулы линейно зависит от расстояния между ядрами. Далее, предположим, что потенциал взаимодействия ядер строго гармонический, т.е. потенциальная энергия пропорциональна квадрату отклонения расстояния между ядрами R от равновесного расстояния Re:  . Здесь
. Здесь  – коэффициент жесткости связи ядер. В дальнейшем отклонение
– коэффициент жесткости связи ядер. В дальнейшем отклонение  будем обозначать как r.
будем обозначать как r.
Как известно, задачу колебания двух материальных точек с массами m1 и m2 можно свести к задаче колебания одной точки с массой 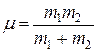 . Масса
. Масса  называется приведенной. Тогда частота колебаний ядер в нашей модели равна
называется приведенной. Тогда частота колебаний ядер в нашей модели равна  . Теперь становится ясным, что простейшей моделью двухатомной молекулы как колебательной системы, является одномерный гармонический осциллятор.
. Теперь становится ясным, что простейшей моделью двухатомной молекулы как колебательной системы, является одномерный гармонический осциллятор.
Для гармонического осциллятора имеется точное решение уравнения Шредингера: известны и волновые функции и соответствующие им значения энергии. Именно, энергетический спектр (набор разрешенных энергий) одномерного гармонического осциллятора задается выражением
 ,
, 
Естественно этой же формулой задается и энергетический спектр двухатомной молекулы в гармоническом приближении.
Здесь уместно сделать замечание относительно размерностей используемых физических величин. В спектроскопии часто используется частота 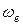 (иначе, волновое число), по определению она равна отношению
(иначе, волновое число), по определению она равна отношению . Частота имеет размерность длины в минус первой степени. Как правило, в литературе используется единица измерения см-1, реже применяется м-1. Кроме того, часто
. Частота имеет размерность длины в минус первой степени. Как правило, в литературе используется единица измерения см-1, реже применяется м-1. Кроме того, часто  формально заменяется на , благодаря чему энергия также принимает размерность см-1. Энергия гармонического осциллятора тогда запишется как
формально заменяется на , благодаря чему энергия также принимает размерность см-1. Энергия гармонического осциллятора тогда запишется как
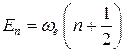 .
.
Можно легко показать (см. раздел 2.1), что в рамках нашей модели поглощение и испускание электромагнитного излучения двухатомными молекулами, состоящими из одинаковых атомов (дипольный момент которых равен нулю) невозможно. Для модели молекул, состоящих из различных атомов (дипольный момент отличен от нуля), существует правило отбора Dn=+1, т.е. с n‑го уровня она может перейти лишь на n-1-й уровень (испуская при этом квант электромагнитного излучения) либо на n+1-й (поглощая квант). Это означает, что если бы данная модель совершенно точно описывала реальную молекулу, то в колебательном спектре поглощения двухатомных молекул была бы лишь одна линия с частотой w, соответствующей разности энергий двух соседних линий:
 .
.
В действительности же молекулы, не имеющие дипольного момента, могут поглощать и испускать излучение за счет отличных от нуля
4 мультипольных моментов: квадрупольного, октупольного, гексадекапольного и т.д. моментов. Обычно интенсивность взаимодействия таких молекул с излучением очень низка, и мы не будем рассматривать этот случай здесь.
Как показывает детальный расчет, для молекул, состоящих из различных атомов, вклад квадрупольного и последующих моментов оказывается пренебрежимо малым, поэтому в дальнейшем мы будем считать, что молекулы имеют только дипольный момент.
В спектрах поглощения реальных молекул, состоящих из разных атомов, помимо интенсивной линии основного тона, имеющей частоту примерно равную w (переход 0→1), имеются и гораздо более слабые линии обертонов с частотами приблизительно 2w, 3w, … (переходы 0→2, 0→3, …). Таким образом, наша простейшая модель описывает реальную молекулу лишь приближенно. Для более точного описания спектров необходимо усложнить нашу модель - ввести понятие ангармоничности.
Обсуждая ангармоничность колебаний молекул, следует отметить, что существует механическая ангармоничность и электрооптическая ангармоничность.
Механическая ангармоничность связана с отличием потенциала взаимодействия ядер молекулы от строго гармонического. Как известно, потенциальная функция реальных молекул имеет минимум при r=0 (равновесное расстояние между ядрами), стремится к бесконечности при стремлении r к 0 (возрастание силы отталкивания при сближении ядер на малое расстояние) и стремится к некоторой постоянной величине при стремлении r к бесконечности. Формально механическая ангармоничность описывается введением разложения потенциальной функции по колебательной координате r:

Коэффициенты 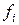 будем называть параметрами механической ангармоничности. При обращении в нуль данных коэффициентов потенциал обращается в гармонический.
будем называть параметрами механической ангармоничности. При обращении в нуль данных коэффициентов потенциал обращается в гармонический.
Механическая ангармоничность приводит к смещению энергетических уровней молекулы от уровней гармонического осциллятора . Зависимость энергетических уровней молекулы En от квантового числа n принято записывать в виде ряда по степеням числа 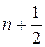 :
:
 (1.1)
(1.1)
Безразмерные коэффициенты  не равны нулю лишь для ангармонических колебаний и поэтому называются параметрами ангармоничности. Перед
не равны нулю лишь для ангармонических колебаний и поэтому называются параметрами ангармоничности. Перед  определен знак минус, потому что для всех молекул данное слагаемое дает отрицательный вклад в энергию и тогда
определен знак минус, потому что для всех молекул данное слагаемое дает отрицательный вклад в энергию и тогда  оказывается положительным, что удобно при вычислениях. Величины параметров ангармоничности быстро убывают с возрастанием номера соответствующего слагаемого: <<1,
оказывается положительным, что удобно при вычислениях. Величины параметров ангармоничности быстро убывают с возрастанием номера соответствующего слагаемого: <<1, 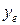 <<…
<<…
Электрооптическая ангармоничность связана с нелинейной зависимостью дипольного момента молекулы от расстояния между ядрами. Данный факт определяется перераспределением электронной плотности в молекуле при изменении межъядерного расстояния. В общем случае, дипольный момент следует записывать в виде степенного разложения по колебательной координате.
Учет механической и электрооптической ангармоничностей позволяет объяснить переходы с Dn≠+1 (обертоны). Следует отметить, что данные переходы объясняются уже при учете хотя бы только одного вида ангармоничности (механической либо электрооптической). В действительности оба вида ангармоничности дают свой вклад в появление обертонов и в их относительные интенсивности (подробнее см. в 3.4).
Наблюдаемые линии в спектрах поглощения соответствуют переходам с основного уровня, так как основной уровень самый заселенный и вероятность данных переходов при поглощении гораздо выше, чем переходов с остальных уровней. При переходе 0→n поглощаются фотоны с энергией, равной разности энергий n–го и основного энергетических уровней:


Если в эксперименте мы можем измерить лишь частоту одной линии, то, разумеется, этого недостаточно, чтобы установить чему равны параметры ангармоничности. По этим данным можно лишь примерно судить о величине частоты 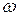 , которую в этом случае можем положить равной экспериментальной частоте. Если наблюдаются две линии, то можно найти гармоническую частоту и параметр
, которую в этом случае можем положить равной экспериментальной частоте. Если наблюдаются две линии, то можно найти гармоническую частоту и параметр 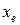 , так как теперь мы имеем два уравнения. Аналогичным образом по трем экспериментальным частотам мы можем найти и параметры и . Заметим, что значение вновь найденной частоты будет несколько отличаться от предыдущей. Тоже справедливо и для . И так далее – чем больше мы имеем экспериментальных частот, тем больше параметров ангармоничности мы можем найти и тем точнее описать потенциал реальной молекулы.
, так как теперь мы имеем два уравнения. Аналогичным образом по трем экспериментальным частотам мы можем найти и параметры и . Заметим, что значение вновь найденной частоты будет несколько отличаться от предыдущей. Тоже справедливо и для . И так далее – чем больше мы имеем экспериментальных частот, тем больше параметров ангармоничности мы можем найти и тем точнее описать потенциал реальной молекулы.