ЗАКОНЫ ЭЛЕКТРОЛИЗА (ЗАКОНЫ ФАРАДЕЯ).
ЭЛЕКТРОХИМИЧЕСКИЕ РЕАКЦИИ.
Электрохимические реакции протекают на границе электрод (проводник первого рода) - электролит (проводник второго рода). Они вызваны невозможностью для электронов - носителей тока в электродах свободно двигаться в электролите. Эти реакции состоят в обмене электронами между электродом и ионами (молекулами) в растворе. На катоде электроны переходят от электрода к иону (или молекуле), на аноде - от иона (молекулы) к электроду, при этом ионы (молекулы) теряют или изменяют свой электрический заряд. Это - первичная электрохимическая реакция, продукты которой нередко вступают в дальнейшие реакции, не связанные непосредственно с переносом тока ионами. Примерами катодных реакций могут служить следующие реакции:
Cu2+ + 2e ® Cu (1)
Fe3+ + e ® Fe2+ (2)
2H3O+ + 2e ® H2(г) + 2H2O (3)
На аноде могут протекать реакции типа:
4OH- ® O2 + 2H2O + 4e (4)
Fe2+ ® Fe3+ + e (5)
2Cl- ® Cl2(г) + 2e (6)
Zn ® Zn2+ + 2e (7)
Материал электрода может участвовать в электрохимической реакции [реакция (7)], но может быть и инертным (остальные реакции). В последнем случае на поверхности электрода могут выделяться металлы [реакция (1)] или газы [реакции (3, 4, 6)]. Наконец, электрохимическая реакция может протекать и при отсутствии перехода ионов из раствора к электроду и обратно. В этих случаях перенос электричества осуществляют только электроны, но у поверхности электрода в растворе ионы изменяют свою валентность [реакции (2) и (5)]. Совокупность двух электрохимическихреакций, из которых одна протекает на катоде, а другая - на аноде, даёт химическую реакцию электролиза или реакцию, протекающую в электрохимическом элементе :
Cu2+ + 2Cl- ® Cu(т) + Cl2 (1) + (6)
2H3O+ + 2OH- ® 1/2 O2 + H2 + 3H2O (3) + 1/2 (4)
или H2O ® 1/2 O2 + H2
Поскольку прохождение электрического тока через электрохимические системы связано с химическими превращениями, между количеством протекающего электричества и количеством прореагировавших веществ должна существовать определенная зависимость. Она была открыта Фарадеем и получила свое выражение в первых количественных законах электрохимии, названных впоследствии законами Фарадея.
Первый закон Фарадея. Количества веществ, превращённых при электролизе, пропорциональны количеству электричества, прошедшего через электролит.
Dm = kэIt = kэq
Dm – количество прореагировавшего вещества; kэ – некоторый коэффициент пропорциональности; q – количество электричества, равное произведению силы тока I на время t. Если q = It = 1, то Dm = kэ , т.е. коэффициент kэ представляет собой количество вещества, прореагировавшего в результате протекания единицы количества электричества. Коэффициент kэ называется электрохимическим эквивалентом.
Второй закон Фарадея отражает связь, существующую между количеством прореагировавшего вещества и его природой : при постоянном количестве прошедшего электричества массы различных веществ, испытывающие превращение у электродов (выделение из раствора, изменение валентности), пропорциональны химическим эквивалентам этих веществ :
Dmi /Ai = const
Можно объединить оба закона Фарадея в виде одного общего закона : для выделения или превращения с помощью тока 1 г-экв любого вещества (1/z моля вещества) необходимо всегда одно и то же количество электричества, называемое числом Фарадея (или фарадеем).
Dm =  It =
It =  It
It
Точно измеренное значение числа Фарадея
F = 96484,52 ± 0,038 к / г-экв
Таков заряд, несомый одним грамм-эквивалентом ионов любого вида. Умножив это число на z (число элементарных зарядов иона), получим количество электричества, которое несёт 1 г-ион. Разделив число Фарадея на число Авогадро, получим заряд одного одновалентного иона, равный заряду электрона:
e = 96484,52 / (6,022035×1023) = 1,6021913×10-19 к
Законы , открытые Фарадеем в 1833 г., строго выполняются для проводников второго рода. Наблюдаемые отклонения от законов Фарадея являются кажущимися. Они часто связаны с наличием неучтённых параллельных электрохимических реакций. Отклонения от закона Фарадея в промышленных установках связаны с утечками тока, потерями вещества при разбрызгивании раствора и т.д.
В технических установках отношение количества продукта, полученного при электролизе, к количеству, вычисленному на основе закона Фарадея, меньше единицы и называется выходом по току.
При тщательных лабораторных измерениях для однозначно протекающих электрохимических реакций выход по току равен единице (в пределах ошибок опыта). Закон Фарадея точно соблюдается, поэтому он лежит в основе самого точного метода измерения количества электричества, прошедшего через цепь, по количеству выделенного на электроде вещества. Для таких измерений используют серебряный или медный, а также йодный и газовый кулонометры (кулонометрия).
Теория электролитов
ТЕОРИЯ ЭЛЕКТРОЛИТИЧЕСКОЙ ДИССОЦИАЦИИ АРРЕНИУСА.Для электролитов понижение температуры замерзания и осмотическое давление значительно больше соответствующих величин для неэлектролитов. В уравнение для осмотического давления p Вант-Гофф ввел эмпирический коэффициент i > 1, физический смысл которого стал понятен с появлением теории электролитической диссоциации :
p = icRT
Теория электролитической диссоциации была предложена Аррениусом (1884-1887), развившим отдельные высказывания ряда ученых.
Основные положения теории Аррениуса :
Соли, кислоты, основания при растворении в воде и некоторых других полярных растворителях частично или полностью распадаются (диссоциируют) на ионы. Эти ионы существуют в растворе независимо от того, проходит через раствор электрический ток или нет. Вследствие этого число независимо движущихся частиц растворенного вещества больше, чем при отсутствии диссоциации, а величины коллигативных свойств растворов возрастают прямо пропорционально числу частиц.
Наряду с процессом диссоциации в растворе идет обратный процесс - ассоциация ионов в молекулы. В качестве меры электролитической диссоциации Аррениус ввел величину степени диссоциации a, определяемую как долю молекул, распавшихся на ионы :
a =  =
= 
Для любой обратимой реакции электролитической диссоциации :
Кn+Аn- Û n+Кz+ + n-Az-
сумма n+ + n- равна общему числу n ионов, образующихся при диссоциации одной молекулы, которое равно коэффициенту Вант-Гоффа i :
 i = 1 + (n+ + n- - 1)×a = 1 + (n - 1)×a
i = 1 + (n+ + n- - 1)×a = 1 + (n - 1)×a
Определив коэффициент i, можно по этому уравнению вычислить степень диссоциации a, если известна величина n.
По мере увеличения разведения коэффициент Вант-Гоффа приближается к простому целому числу (2, 3, 4 - в зависимости от числа ионов, образующихся из одной молекулы вещества).
Диссоциация растворенных веществ на ионы подчиняется тем же законам химического равновесия, что и др. реакции :
Кд = 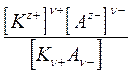
где Кд - константа диссоциации, выраженная через концентрации.
Диссоциация сильных электролитов равна 100% или почти 100%, так что концентрации ионов можно считать равными молярности растворенного вещества, умноженной на z. При диссоциации слабого электролита устанавливается равновесие между недиссоциированными молекулами и ионами. Рассмотрим простейший пример, когда молекула распадается только на два иона :
СН3СООН Û СН3СОО- + Н+
с - aс aс aс
Кд =  =
=  ; Кс =
; Кс = 
Последнее равенство является простейшей формой закона разведения Оствальда (1888). Чем больше Кс, тем выше степень диссоциации. Т.о., величина Кс может служить мерой силы кислоты, т.е. мерой кислотности. Для электролитов средней силы (Н3РО4 - первая ступень, Са(ОН)2, СНСl2СООН) значения Кс лежат в пределах от 10-2 до 10-4; для слабых электролитов (СН3СООН, NН4ОН) Кс = 10-5 - 10-9; при Кс < 10-10 электролит считается очень слабым (Н2О, С6Н5ОН, С6Н5NН2, НСN).
Если a очень мала, то ее величиной можно пренебречь по сравнению с 1, и формула примет вид :
Кс = a2с ; a = 
Если электролит распадается больше чем на два иона, то зависимость Кс от a усложняется :
СаCl2 Û Ca2+ + 2Cl-
с (1- a) aс 2aс
Кс =  =
=  =
= 
При малой a : a = 
Величина Кс является постоянной только для очень разбавленных растворов, коэффициенты активности которых можно считать равными 1. Вообще же Кс (как и a) зависит от концентрации раствора; Кс иногда еще называют классической константой диссоциации.
Кс (a) зависит также от температуры : зависимость Кс слабых кислот в воде проходит через максимум. Это можно объяснить влиянием двух противоположно направленных воздействий. С одной ст., всякая диссоциация протекает с поглощением тепла, и, следовательно, при повышении Т равновесие должно смещаться в сторону большей диссоциации. С др. ст., при повышении Т диэлектрическая проницаемость воды, служащей растворителем, уменьшается, а это способствует воссоединению ионов. Кс максимальна при той Т, при которой влияние второго фактора начинает преобладать. Обычно изменение Кд с повышением Т невелико.