АТЕРОСКЛЕРОЗ
Лекция 5
• Атеросклероз (от греч. athere — кашица и sclerosis — уплотнение) — хроническое заболевание, возникающее в результате
нарушения липидного и белкового обмена, характеризующееся поражением артерий эластического и мышечно-эластического типа в виде очагового отложения во внутренней оболочке липи-дов и белков и реактивного разрастания соединительной ткани.
Следует отличать атеросклероз от артериосклероза, которым обозначают склероз артерий независимо от причины и механизма развития. Атеросклероз — лишь наиболее частая разновидность артериосклероза, отражающая нарушение метаболизма липидов и белков (метаболический атеросклероз). В таком толковании термин "атеросклероз" был введен в 1904 г. Маршаном и обоснован экспериментальными исследованиями Н.Н.Аничкова.
Атеросклероз широко распространен среди населения экономически развитых стран Европы и Северной Америки, в которых связанная с ним патология (ишемическая болезнь сердца, цереброваскулярные болезни и пр.) вышла на первое место среди причин смертности. Во второй половине XX века атеросклероз приобрел характер эпидемии — стал быстро распространяться в географические зоны, в которых раньше не наблюдался — Японию, Китай, некоторые африканские страны. Однако по-прежнему смертность от атеросклероза в различных странах подвержена значительным колебаниям (в Финляндии в 10 раз выше, чем в Японии), имеются страны и отдельные популяции населения, для которых атеросклероз является исключительной редкостью. Все это не позволяет рассматривать атеросклероз как фатальную неизбежность, естественное следствие жизни и старения человека. Раскрытие причин атеросклероза, механизмов его развития является важнейшей проблемой медицины.
Этиология.В настоящее время общепризнано, что атеросклероз — полиэтиологическое заболевание, связанное с влиянием различных экзогенных и эндогенных факторов, среди которых основное значение имеют наследственные, средовые и пищевые. При различных формах атеросклероза роль отдельных факторов варьирует. Так, у людей с семейными наследственными формами раннего атеросклероза на первый план выступают генетические факторы, тогда как массовое распространение атеросклероза связано главным образом с факторами среды и особенностями питания. Часто отмечается сочетание различных факто-' ров, причем некоторые присоединяются в ходе развития заболевания. Поэтому при атеросклерозе сложно разграничить этиологические и патогенетические факторы.
Эпидемиология.Массовые эпидемиологические обследования населения различных стран позволили выявить ряд факторов, влияющих на частоту атеросклероза, — факторы риска. Не подвергается сомнению значение возраста, пола и семейной предрасположенности. Среди прочих факторов основными являются: гиперлипидемия (гиперхолестеринемия), артериальная гипертензия, курение, сахарный диабет. Кроме того, прослеживается зависимость между выраженностью атеросклероза и стрессовыми ситуациями, малоподвижным образом жизни, тучностью, гиперурикемией.
Возраст. Увеличение частоты и выраженности атеросклероза с возрастом — факт неоспоримый. Это позволило некоторым исследователям придавать основное значение в развитии атеросклероза возрастным изменениям сосудистой стенки и рассматривать атеросклероз не как болезнь, а как гериатрическую проблему.
Пол. Во всех возрастных группах больных атеросклерозом преобладают мужчины. Различия становятся менее явными в постклимактерическом периоде. После 70 лет различия нивелируются.
Семейная предрасположенность. При атеросклерозе семейная предрасположенность имеет большое значение. Часто она объясняется наличием других (одного или нескольких) генетически обусловленных факторов риска — сахарного диабета, артериальной гипертензии, гиперлипидемии. Однако иногда семейная предрасположенность является единственным обнаруживаемым у больных атеросклерозом фактором риска.
Гиперлипидемия (гиперхолестеринемия). Большинством авторов гиперлипидемия признается как ведущий фактор риска. Однако в последнее время стали придавать значение не столько повышению уровня холестерина в крови, сколько нарушению соотношения между липопротеидами низкой (ЛПНП) и очень низкой плотности (ЛПОНП) — атерогенными, и липопротеидами высокой плотности (ЛПВП) — антиатерогенными. В норме это соотношение составляет 4:1 и значительно возрастает при атеросклерозе. Результаты обширных эпидемиологических исследований позволили установить, что 2/3 случаев атеросклероза обусловлены нарушением обмена ЛПНП и ЛПОНП, в V3 наблюдений развитие атеросклероза объясняют снижением уровня ЛПВП.
О значении гиперлипидемии при атеросклерозе свидетельствуют следующие факты:
▲ как правило, в популяциях с высоким содержанием холестерина в крови высока распространенность атеросклероза. Так, при повышении уровня холестерина в крови до 265 мг/л (верхняя граница нормы равна 20 мг/л) в 5 раз возрастает риск развития ишемической болезни сердца (ИБС). При снижении — результат обратный. Еще более существенны корреляции с уровнем ЛПНП;
▲ гиперлипидемия приводит к атеросклерозу вне зависимости от ее происхождения (является ли она наследственной или приоб-
ретенной, связанной с экзогенными или эндогенными причинами). Так, генетически обусловленная семейная гиперхолестеринемия приводит к развитию раннего атеросклероза с частыми клиническими проявлениями в детском возрасте. Для ряда приобретенных синдромов, сопровождающихся гиперлипидемией (нефротический синдром, гипотиреоз), также характерен выраженный атеросклероз. Имеются доказательства того, что атеросклероз часто развивается в связи с гиперхолестеринемией, обусловленной избыточным потреблением холестерина и насыщенных жирных кислот;
▲ в эксперименте атеросклероз воспроизводится при скармливании животным холестерина [холестериновая модель атеросклероза Н.Н.Аничкова (1913) положила начало биохимическому этапу в изучении атеросклероза];
▲ при анализе атеросклеротической бляшки выявляется 10-кратное по сравнению с нормальной внутренней оболочкой содержание липидов, в основном за счет линолеатов, извлекаемых из ЛПНП.
Для понимания значения гиперлипидемии при атеросклерозе необходимо представить современный взгляд на метаболизм липидов в организме (схема 35). Липиды циркулируют в крови в виде липопротеидных комплексов, которые содержат холестерин, холестеринэстеры, триглицериды, фосфолипиды и белки — апопротеины (апо). Апопротеины обеспечивают определенную структуру липопротеидной частицы, направленный транспорт липидов в организме, высокоаффинное их связывание с клеточными рецепторами — апорецепторами. Известно 13 апопротеинов, выполняющих различную функцию. Наибольшее значение имеют апоА-1, апоС-2, апоЕ, апоВ-48, апоВ-100.
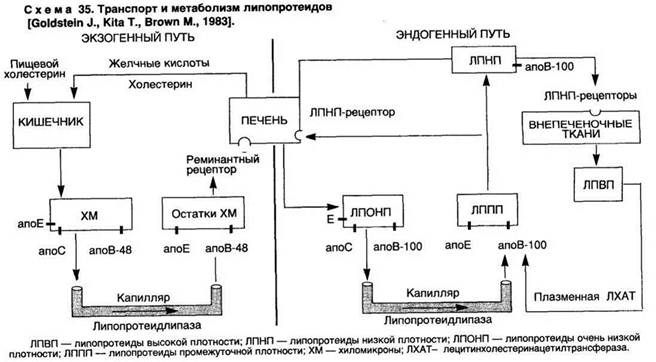
Циркулирующие липопротеиды имеют различную величину и плотность, которая определяется различным соотношением в них белков и липидов. В зависимости от плотности выделяют 5 классов липопротеидов: хиломикроны (ХМ), липопротеиды очень низкой плотности (ЛПОНП), липопротеиды промежуточной плотности (ЛППП), липопротеиды низкой плотности (ЛПНП) и липопротеиды высокой плотности (ЛПВП).
1. ХМ — наиболее крупные частицы. Образуются в энтероцитах тонкой кишки. Содержат преимущественно экзогенные (пищевые) триглицериды, небольшое количество холестерина и апо-48 (основной апопротеин, осуществляющий транспорт экзогенных липидов из кишки в кровь), а также апоЕ, апоС.
2. ЛПОНП состоят преимущественно из триглицеридов и холестерина (содержат 10—15 % циркулирующего холестерина), содержат апоЕ, апоС и апоВ-100. Осуществляют транспорт липидов, которые синтезируются в печени.
3. ЛППП образуются из ЛПОНП, состоят примерно из равного количества холестерина (преимущественно эфиров) и триглицеридов, а также апоВ-100 и апоЕ.
4. ЛПНП состоят преимущественно из эфиров холестерина и апоВ-100. Переносят около 70 % циркулирующего холестерина. Отмечается наиболее выраженная корреляция с атеросклерозом. Еще большей атерогенностью обладают модифицированные ЛПНП — оксилированные, ацетилированные, а также липопротеид-а (образующийся при приеме богатой холестерином пищи), и комплексы ЛПНП с иммуноглобулинами, образующиеся при гиперлипидемии.
5. ЛПВП — самые мелкие частицы, содержат 20—25 % циркулирующего холестерина в виде его эфиров с ненасыщенными жирными кислотами, фосфолипиды и апоА (ключевой апопротеин ЛПВП), апоС и апоЕ. Осуществляют "откачку" излишков холестерина из клеток в виде эфиров и передачу его на ЛППП. Кроме того, в печеночных клетках ЛПВП стимулируют выведение холестерина в виде желчных кислот.
Экзогенный и эндогенный холестерины транспортируются различными путями. Пищевые триглицериды и холестерин в энтероцитах тонкой кишки собираются в ХМ, содержащие апоВ-48. В крови из ХМ под воздействием липопротеидлипазы эндотелия сосудов (фермент активируется под воздействием апоС) освобождаются жирные кислоты и глицерин, которые поступают в жировые и мышечные клетки, где окисляются или вновь включаются в синтез триглицеридов. Остатки ХМ (ремнанты) прикрепляются к апоЕ-рецепторам печеночных клеток, подвергаются эндоцитозу и затем разрушаются в лизосомах. Таким образом, экзогенные триглицериды утилизируются в жировой и мышечной ткани, а экзогенный холестерин переносится в печень, где синтезируются ЛПОНП, содержащие триглицериды и холестерин, а также апоЕ, апоС, но вместо азоВ-48 — апоВ-100. В крови под воздействием липопротеидлипазы ЛПОНП превращаются в короткоживущие ЛППП (в крови здоровых людей они не обнаруживаются), часть из которых захватывается апоВ, Е-рецепторами печеночных клеток. Большая часть ЛППП после обогащения эфирами холестерина, образующимися под воздействием плазменной лецитинхолестеринацетилтрансферазы (ЛХАТ) на ЛПВП (активация фермента происходит под действием апоА-2 — основного апопротеина ЛПВП), превращаются в ЛПНП, которые состоят из эфиров холестерина и одного апопротеина — апоВ-100. ЛПНП являются основными поставщиками эндогенного холестерина в клетки.
Существует два пути доставки эндогенного холестерина в клетки: ЛПНП-рецепторный регулируемый и вне ЛПНП-рецепторный нерегулируемый эндоцитоз:
ЛПНП-рецепторный регулируемый эндоцитоз. В норме большая часть (более 2/3) ЛПНП удаляется из крови и утилизируется клетками с помощью ЛПНП-рецепторов, которые имеются как на печеночных, так и внепеченочных клетках (надпочечники, фибробласты, гладкие мышечные клетки, лимфоциты, эндотелий и др.); 50—70 % ЛПНП утилизируются печенью. ЛПНП-ре-цептор — трансмембранный гликопротеид, который осуществляет связь клеток с липопротеидами, имеющими апоВ- и апоЕ-лиганды (ЛПНП и ЛППП), с последующим эндоцитозом и гидролизом в лизосомах. При этом освободившиеся рецепторы возвращаются в клеточную мембрану. В периферических клетках здоровых людей ЛПНП-рецепторы при загрузке лигандом автоматически блокируют синтез холестерина в клетке (ингибиторами являются метаболиты холестерина, которые возникают при активации ЛПНП-рецепторов). ЛПНП-рецепторный регулируемый эндоцитоз — механизм, с помощью которого клетки контролируют свою потребность в холестерине, необходимом прежде всего для синтеза мембран. При уменьшении уровня внутриклеточного холестерина или снижении уровня ЛПНП в крови освобождается больше рецепторов и наоборот. Неэстерифицирован-ный холестерин, высвобождающийся при обычном обновлении мембран, извлекается из клеток с помощью ЛПВП либо в водной фазе по градиенту концентрации, либо через ЛПВП-рецепторы (более сложный путь).
Вне ЛПНП-рецепторный нерегулируемый эндоцитоз. Меньшая часть ЛПНП утилизируются клетками, минуя ЛПНП-рецепторы. Нерегулируемый (т.е. ненасыщаемый) эндоцитоз осуществляется в основном клетками моноцитарно-макрофагальной (ретикуло-эндотелиальной) системы, в которых этот путь преобладает над ЛПНП-рецепторным. Эндотелиальная клетка, макрофаг способны захватывать липопротеиды, модифицированные-липопротеиды (оксидированные, ацетилированные) из крови с помощью рецепторов к модифицированным ЛПНП — скэвенд-жер-рецепторов (рецепторы "клеток-мусорщиков"). Кроме того, с помощью рецепторов к Fc-фрагментам эти клетки способны захватывать иммунные комплексы, содержащие липопротеиды, а с помощью Э-ЛПОНП-рецепторов — модифицированные ЛПОНП. Излишки холестерина, накапливающегося в лизосомах, макрофаг способен выводить с помощью сложного механизма — ретроэндоцитоза ЛПВП, состоящего из внутриклеточного захвата — эндоцитоза ЛПВП, обогащения их холестерином и экзоцитоза — выброса из клетки. Таким образом, в норме баланс внутриклеточного холестерина в клетках макрофагальной системы определяется не только потоком липопротеидных частиц в клетку, но и механизмами его обратного транспорта.
Значение нерецепторного нерегулируемого пути выведения ЛПНП резко возрастает при гиперлипидемии, когда блокируется большая часть ЛПНП-рецепторов и образуются модифицированные ЛПНП. Нерегулируемый захват ЛПНП (а также модифицированных fJ-ЛПОНП) в этих условиях приводит к несостоятельности систем выведения холестерина, излишнему накоплению его и образованию пенистых, или ксантомных, клеток (от греч. xantos — желтый), с которыми связан атерогенез. Вот почему ЛПНП и ЛПОНП называют атерогенными липопротеидами.
Гиперлипидемии чрезвычайно разнообразны. Они могут быть обусловлены первичными генетическими нарушениями в системе метаболизма липидов — первичные гиперлипидемии либо другими заболеваниями (сахарный диабет, нефротический синдром) — вторичные гиперлипидемии. Первичные гиперлипидемии могут быть обусловлены дефектом одного гена или полигенным. Учитывая, что только транспорт липидов регулируется 100 генами (кодирующими апорецепторы, апопротеины и их лиганды, ферменты и пр.), можно представить все многообразие возможных форм первичных гиперлипидемии. В настоящее время в зависимости от увеличения уровня тех или иных липопротеидов выделено пять типов семейных гиперлипидемии (гиперхолестеринемий), характеризующихся различным атерогенным потенциалом. Наиболее хорошо изучена первичная семейная гипер-3-липопротеидемия (тип 2А), связанная с наследственным дефектом ЛПНП-рецептора. Болезнь проявляется полной или частичной утратой способности клеток (прежде всего гепатоцитов) удалять из кровотока ЛПНП, что приводит к значительному увеличению их уровня в крови и высокому риску развития атеросклероза, часто в детском возрасте (у гомозигот смерть от инфаркта миокарда наступает в возрасте от 3 до 33 лет). Исследование этого типа семейной гиперлипидемии, выяснение роли ЛПНП-рецепторов в ее генезе явились важной вехой в изучении атеросклероза. Неудивительно, что за доказательство рецепторной теории некоторых форм ускоренного атеросклероза американские исследователи И.Гольдштейн и М.Браун в 1985 г. были удостоены Нобелевской премии.
По-видимому, роль ЛПНП-рецепторов в развитии атеросклероза универсальна. При наследственных гиперлипидемиях дефицит ЛПНП-рецепторов первичен, при других же состояниях он может быть вторичным и подключаться в качестве патогенетического фактора. Так, любая гипер-Э-липидемия (в том числе связанная с злоупотреблением богатой холестерином и насыщенными жирными кислотами пищей) приводит (как это указывалось ранее) к снижению экспрессии ЛПНП-рецепторов и нерегулируемому клеточному эндоцитозу, что повторяет события при
наследственных дефектах ЛПНП-рецепторов. Существует мнение, что увеличивающийся с возрастом риск развития атеросклероза также связан с приобретаемыми качественными и количественными дефектами ЛПНП-рецепторов, что в свою очередь может привести к гиперлипидемии.
Артериальная гипертензия. Вне зависимости от ее генеза является одним из основных факторов риска при атеросклерозе. Наиболее четко коррелирует выраженность атеросклероза с уровнем диастолического давления. Значительно возрастает роль артериальной гипертензии с возрастом. Как считают некоторые исследователи, у людей старше 45 лет с артериальной гипертензией связан больший риск развития атеросклероза, чем с гиперлипидемией. Значение этого фактора риска подтверждается нередким появлением атеросклеротических изменений в "нетипичных" для него сосудах при наличии местной гипертензии — в легочной артерии при гипертензии малого круга, в воротной вене при портальной гипертензии.
Курение. У людей, курящих 1—2 пачки сигарет в день, смерть от атеросклероза регистрируется почти в 2 раза чаще, чем у некурящих. Особенно важен этот фактор для развития атеросклероза венечных артерий и связанной с ним ишемической болезни сердца.
Сахарный диабет. Атеросклероз (макроангиопатия) — одно из основных проявлений сахарного диабета. Нарушение обмена веществ при сахарном диабете сопровождается гиперлипидемией с появлением большого количества модифицированных ЛПНП (преимущественно гликозилированных), обладающих наибольшей атерогенностью. Особенно часто возникает гангрена ног, обусловленная облитерирующим атеросклерозом.
Стрессовые ситуации. Нервному фактору — стрессовым и конфликтным ситуациям, с которыми связано психоэмоциональное перенапряжение, придается большое значение в раз-' витии атеросклероза, поэтому атеросклероз рассматривается как болезнь сапиентации.
Результаты статистических исследований показали, что сочетание различных факторов риска значительно усиливают проявления атеросклероза. Однако у некоторых больных атеросклерозом факторы риска не выявляются и, наоборот, при наличии даже нескольких факторов проявления бывают минимальными. Поэтому патогенез атеросклероза по-прежнему остается предметом дискуссии.
Патогенез. Безуспешные, более чем вековые попытки найти первопричину атеросклероза и создать единую универсальную схему его патогенеза не увенчались успехом и привели во второй половине XX века к признанию полиэтиологической природы этого заболевания. Ни одна из предложенных теорий патогенеза атеросклероза не смогла полностью раскрыть механизмы заболевания и определить роль всех факторов риска. Недостатком большинства теорий является то, что они часто рассматривают не патогенез заболевания в целом, а механизмы развития бляшки (атерогенез) вне связи с общими метаболическими нарушениями. Между тем именно первичные биохимические нарушения в большинстве случаев являются главной движущей силой заболевания.
Представления о ведущих факторах риска атеросклероза сформулированы в тромбогенной, иммунной, клональной, вирусной теориях, теории реакции на повреждение, липопротеидной и нервно-метаболической гипотезах. Наибольшего внимания заслуживают липопротеидная теория и теория реакции на повреждение как наиболее полно отражающие совокупность полученных современных данных.
В основе липопротеидной теории лежит имбибиционная теория Р.Вирхова, рассматривавшего атеросклеротические бляшки как воспалительную реакцию на инфильтрацию сосудистой стенки плазменными компонентами, и в дальнейшем преобразованная в инфилътрационную теорию Н.Н.Аничкова, экспериментально доказавшего роль гиперхолестеринемии при атеросклерозе. Скармливание животным холестерина приводит к гиперхолестеринемии, отложению холестерина и его эфиров в стенке аорты и артерий, развитию атеросклеротических изменений. Блестящим обоснованием липопротеидной теории явилась рецепторная теория некоторых наследственных форм атеросклероза И.Гольдштейна и М.Брауна. Ключевым моментом патогенеза атеросклероза, согласно липопротеидной теории, являются нарушение систем, обеспечивающих синтез и катаболизм липопротеидов, с развитием гиперлипидемии, образованием модифицированных ЛПНП и ЛПОНП и перевод регулируемого рецепторного процесса захвата липопротеидов на нерегулируемый. Атеросклероз, согласно этой теории, следует рассматривать как реакцию сосудистой стенки на появление модифицированных липопротеидов. Инициирующим моментом при этом служит нерегулируемый их захват неповрежденным эндотелием. Слабым звеном этой теории является невозможность объяснить те случаи атеросклероза, при которых отсутствует атерогенная гиперлипидемия.
Теория реакции на повреждение, как наиболее полно отражающая все стороны патогенеза, находит наибольшее число сторонников. В качестве инициального фактора атерогенеза (возникновения атеросклеротической бляшки) рассматривается повреждение сосудов, которое может быть вызвано самыми разнообразными факторами: гиперлипидемией, механическим воздействием, стрессом, иммунными механизмами, токсинами, вирусами или другими инфекционными агентами, гемодинамическими факторами (гипертензией, повторными спазмами, неправильными турбулентными потоками крови в области ветвления сосудов и т.д.). Преимущества этой теории в том, что она логично объясняет роль различных факторов риска в патогенезе как инициаторов хронического повреждения сосудов; допускает вероятность минимальных повреждений эндотелия как триггера процесса; особое и наибольшее значение как повреждающему фактору придает гиперлипидемии и модифицированным ЛПНП, может объяснить атерогенез при отсутствии последних: в этой ситуации основное значение придается инссудации с последующим образованием модифицированных липопротеидов во внутренней оболочке сосуда. Теория реакции на повреждение находит многочисленные экспериментальные подтверждения.
Согласно тромб ore иной теории Дж.Б.Дьюгеда (прототипом этой теории следует считать инкрустационную теорию К.Рокитанского), атеросклеротическая бляшка является следствием организации образующихся в сосудах (возможно, повторно) пристеночных и интрамуральных тромбов. Теория основывается на некотором морфологическом сходстве организованных тромбов и атеросклеротических бляшек; довольно частом обнаружении фибрина и тромбоцитов как на поверхности сосудистого эндотелия, так и в атеросклеротических бляшках; на клинических и экспериментальных исследованиях, показывающих, что гиперлипидемия вызывает уменьшение времени свертывания крови и нарушение фибринолиза. В настоящее время роль тромбоцитов в инициации атеросклероза представляется малообоснованной.
Нервно-метаболическая теория А.Л.Мясников а исключительную роль в развитии атеросклероза отводит нервному фактору — стрессовым и конфликтным ситуациям, с которыми связано психоэмоциональное перенапряжение, ведущее к нарушению нейроэндокринной регуляции белково-липидного обмена и вазомоторным расстройствам.
Сторонники моноклональной теории считают, что в основе атерогенеза лежит опухолевая пролиферация гладких мышечных клеток (ГМК), аналогичная таковой в лейомиомах. В качестве аргумента приводятся данные о том, что пролифериру-ющие ГМК, обнаруживаемые в атеросклеротической бляшке, относятся (как и опухолевые) к одному клону. Сторонники этой теории не исключают, что в качестве митогена могут выступать модифицированные ЛПНП, вирусы.
Иммунологическая теория во многом основывается на результатах клинико-морфологических и экспериментальных исследований отечественных ученых — А.Н.Климова, В.А.На-горнева и др. Эти результаты позволили прийти к заключению, что изменения в сосудистой стенке, возникающие на ранних стадиях атеросклероза, есть не что иное, как иммунное воспаление. Выявленные при атеросклерозе в крови и сосудистой стенке аутоиммунные комплексы, в состав которых входят липопротеиды, а также характерные изменения в иммунокомпетентных органах (значительное увеличение числа лимфобластов в периартериальной зоне селезенки и паракортикальной зоне лимфатических узлов, резкое возрастание в В-зоне белой пульпы селезенки и коре лимфатических узлов вторичных лимфоидных фолликулов со светлыми центрами и выраженной плазматизацией) и стенке артерий в зоне отложения липидов (60 % всех обнаруживаемых клеток составляют макрофаги, часто имеющие секреторный фенотип, а 20 % — Т-лимфоциты, преимущественно хелперы; обнаружены многочисленные цитокины — ИЛ-1, ИЛ-2, ИЛ-6,7-ин-терферон и др.) позволили выделить особую нозологическую форму атеросклероза, при которой развивающееся иммунопатологическое состояние имеет решающее значение в генезе заболевания. Иммунологическую гипотезу атеросклероза подтверждают многочисленные наблюдения прогрессирующего атеросклероза у пациентов, которым была произведена аллотрансплантация различных органов — сердца, печени, почек, сосудов. Отмечено ускоренное развитие атеросклероза не только в сосудах донорского органа, но и в собственных артериях больного. Иммунодепрессанты частично блокируют развитие процесса. Экспериментально подтверждена роль иммунных комплексов в повреждении эндотелия и формировании пенистых клеток.
Вирусная теория основана на следующих фактах: цитомегаловирусная инфекция, нередко возникающая после трансплантации сердца, может приводить к усилению атеросклеротических проявлений; в стенке артерий иногда обнаруживается вирус герпеса (как в пораженных, так и непораженных участках); атеросклероз может быть воспроизведен в эксперименте с помощью V.herpes у животных с нормальным уровнем холестерина; заражение цыплят вирусом Марека приводит к развитию у них атеросклеротических бляшек; в инфицированной V.herpes культуре ГМК наблюдается накопление свободного и эстерифицированного холестерина; V.herpes индуцирует синтез цитокинов, действующих как промоторы факторов роста для сосудистых клеток. Механизм действия вирусов объясняют связыванием вируса с эндотелиальными клетками и ГМК посредством их рецепторов к фактору роста фибробластов, а также с выраженным цитотоксическим эффектом на эндотелий и ГМК, который запускает механизмы атерогенеза.
Различные теории патогенеза в настоящее время рассматриваются не как взаимоисключающие, а как дополняющие друг друга и более детально раскрывающие возможные механизмы развития различных форм атеросклероза.
Итак, патогенез атеросклероза, учитывая его многофакторность, можно представить следующим образом (схема 36).

1. В большинстве случаев развитию атеросклероза предшествует атерогенная дислипопротеидемия, сопровождающаяся появлением модифицированных липопротеидов, которые усиленно захватываются эндотелиальными клетками (с помощью рецепторов к Р-ЛПОНП и скавенджер-рецепторов) и переносятся в субэндотелиальное пространство.
2. Центральным звеном атерогенеза является повреждение эндотелия (модифицированными липопротеидами или любыми другими факторами — вирусами, иммунными комплексами, бактериальными токсинами и т.д.), повышение сосудистой проницаемости и инссудация плазменных компонентов, в том числе липопротеидов, во внутреннюю оболочку сосудов.
3. Поврежденный эндотелий экспрессирует адгезивные молекулы, что приводит к прилипанию (адгезии) тромбоцитов и моноцитов. Последние в большом количестве начинают проникать во внутреннюю оболочку сосудов, где превращаются в макрофаги и продуцируют многочисленные цитокины, усиливающие клеточную пролиферацию и миграцию — интерлейкин-1 (ИЛ-1), фактор некроза опухоли (ФНО), тромбоцитарный фактор роста (ТцФР) и др.
4. ГМК под влиянием ТцФР (который, помимо макрофагов, продуцируют также эндотелий и сами ГМК) принимают синтетический фенотип (обычно преобладает сократительный фенотип), мигрируют во внутреннюю оболочку сосудов, пролиферируют, синтезируют коллагеновые, эластические волокна, про-теогликаны, т.е. создают основу атеросклеротической бляшки.
5. Липопротеиды во внутренней оболочке сосудов подвергаются дальнейшей модификации (преимущественно пероксидации под воздействием факторов, вырабатываемых макрофагами), образуют комплексы с протеогликанами, в таком виде захватываются макрофагами, которые при истощении систем утилизации и выведения (прежде всего лизосом) загружаются липидами и превращаются в ксантомные клетки. Часть ксантомных клеток образуются из ГМК, которые, обладая рецепторами к модифицированным Э-ЛПОНП, нерегулируемо поглощают их.
6. Последующие изменения бляшки связаны с новообразованием в ней капилляров под воздействием ФР, привлечением других клеточных элементов — Т- и В-лимфоцитов, фибробластов; некрозом центральных отделов, склерозом, гиалинозом, обызвествлением.
Патологическая анатомия и морфогенез.Основным морфологическим выражением атеросклероза является бляшка, сущность которой хорошо отражает название болезни: в центре — липидно-белковый детрит — (athere), вокруг — разрастание соединительной ткани — склероз. Обычно поражаются артерии эластического (аорта) и мышечно-эластического типа (крупные органные артерии), значительно реже в процесс вовлекаются мелкие артерии мышечного типа.
Атеросклеротический процесс проходит определенные стадии (фазы), которые имеют макроскопическую и микроскопическую характеристику (морфогенез атеросклероза — атерогенез).
При макроскопическом исследовании различают следующие виды атеросклеротических изменений, отражающих динамику процесса:
▲ жировые пятна и полоски;
▲ фиброзные бляшки;
▲ осложненные поражения, представленные фиброзными бляшками с изъязвлением, кровоизлияниями и наложениями тромботических масс;
▲ кальциноз или атерокальциноз.
Жировые пятна и полосы — это участки желтого или желто-серого цвета (пятна), которые иногда сливаются и образуют полоски, но не возвышаются над поверхностью внутренней оболочки сосуда. Они содержат липиды, выявляемые при тотальной окраске сосуда красителями на жир, например Суданом. Раньше всего жировые пятна и полоски появляются в аорте на задней стенке и у места отхождения ее ветвей, позже — в крупных артериях.
Жировые пятна обнаруживаются у всех детей вне зависимости от географической зоны проживания, пола и расы. В первой декаде жизни площадь их равна примерно 10 %, в третьей — 30—50 % от общей площади сосуда. Вопрос о связи полос с атеросклеротическими бляшками остается открытым, поскольку жировые полосы встречаются в местах, где бляшек обычно не бывает. Кроме того, полосы обнаруживаются у народов, для которых другие атеросклеротические проявления нехарактерны. Однако в венечных артериях локализация жировых полос и атеросклеротических бляшек совпадает, поэтому предполагают, что только часть пятен и полос прогрессирует в бляшки при наличии предрасполагающих к атеросклерозу факторов.
Фиброзные бляшки — плотные овальные или округлые, белые или желто-белые образования, содержащие липиды и возвышающиеся над поверхностью внутренней оболочки сосуда. Они часто сливаются между собой, придают внутренней поверхности бугристый вид и приводят к сужению просвета сосуда (стенозирующий атеросклероз).
Распределение бляшек довольно постоянно и отличается от распределения пятен. Наиболее часто бляшки располагаются в брюшной аорте, в артериях сердца, мозга, почек, нижних конечностей, сонных артериях и др. Чаще поражаются те участки сосудов, которые испытывают гемодинамическое (механическое)
воздействие — в области ветвлений и изгибов артерий, на стороне их стенки, которая имеет жесткую подстилку (аргумент в пользу патогенетической теории реакции на повреждение).
Осложненные поражения возникают в тех случаях, когда в толще бляшки преобладает распад липидно-белковых комплексов и образуется детрит, напоминающий содержимое ретенционной кисты сальной железы, т.е. атеромы. Поэтому такие изменения называют атероматозными. Прогрессировать атероматозных изменений ведет к деструкции покрышки бляшки, ее изъязвлению (атероматозная язва), кровоизлияниям в толщу бляшки (интрамуральная гематома) и образованию тромботических наложений на месте изъязвления бляшки. С осложненными поражениями связаны острая закупорка артерии тромбом и развитие инфаркта, эмболия как тромботическими, так и атероматозными массами, образование аневризмы сосуда в месте его изъязвления, а также артериальное кровотечение при разъедании стенки сосуда атероматозной язвой.
Кальциноз, или атерокальциноз, — завершающая фаза атеросклероза, которая характеризуется отложением в фиброзные бляшки солей кальция, т.е. их обызвествлением. Бляшки приобретают каменистую плотность (петрификация бляшек), стенка сосуда в месте петрификации резко деформируется.
Различные виды атеросклеротических изменений нередко сочетаются: в одном и том же сосуде, например в аорте, можно видеть одновременно жировые пятна и полосы, фиброзные бляшки, атероматозные язвы с тромбами и участками атерокальци-ноза, что свидетельствует о волнообразном течении атеросклероза.
Микроскопическое исследование позволяет уточнить и дополнить характер и последовательность развития изменений, свойственных атеросклерозу. На основании его результатов выделены следующие стадии морфогенеза атеросклероза: 1) долипидная; 2) липоидоз; 3) липосклероз; 4) атероматоз; 5) изъязвление; 6)атерокальциноз.
Долипидная стадия характеризуется изменениями, отражающими общие нарушения метаболизма при атеросклерозе, повышение проницаемости и повреждение внутренней оболочки сосуда. В эндотелиальных клетках появляются липидные капли, иногда занимающие до 50 % цитоплазмы. Характерны отек эндотелиальных клеток, исчезновение гликокаликса, повреждение ци-толеммы, раскрытие межэндотелиальных контактов, появление в субэндотелиальном слое капель жира, белков плазмы, фибриногена (фибрина). Уже в ранние стадии можно наблюдать пролиферацию ГМК и макрофагов, в некоторых случаях еще до выраженных повреждений эндотелия и появления липидов и белков. По-видимому, с синтезом ГМК протеогликанов можно связать развивающийся мукоидный отек внутренней оболочки сосудов,
часто являющийся наиболее ранним микроскопическим проявлением. Переход морфогенеза в следующую стадию, по-видимому, следует расценивать как истощение и несостоятельность систем, обеспечивающих выведение из внутренней оболочки липопротеидов и других метаболитов. Прежде всего это связано с несостоятельностью макрофагов.
В стадии липоидоза отмечается очаговая инфильтрация внутренней оболочки сосуда, особенно поверхностных ее отделов, липидами (холестерином), липопротеидами, белками, что приводит к образованию жировых пятен и полос. Липиды диффузно пропитывают внутреннюю оболочку и накапливаются в ГМК и макрофагах, которые превращаются в ксантомные клетки. Выражены набухание и деструкция эластических мембран.
Липосклероз характеризуется разрастанием соединительнотканных элементов внутренней оболочки сосудов в участках отложения и распада липидов и белков, что приводит к формированию фиброзной бляшки. В краях бляшки происходит новообразование тонкостенных сосудов, которые также становятся дополнительным источником поступления липопротеидов и плазменных белков.
При атероматозе липидные массы, составляющие центральную часть бляшки, а также прилежащие коллагеновые и эластические волокна распадаются. При этом образуется аморфная масса, в которой обнаруживаются кристаллы холестерина (атероматозный детрит). В краях бляшки определяются многочисленные сосуды, врастающие из vasa vasorum, а также ксантомные клетки, лимфоциты, плазматические клетки. Атероматозные массы отграничены от просвета сосуда слоем зрелой, иногда гиалинизированной соединительной ткани (покрышка бляшки). Мышечная оболочка часто атрофируется, иногда подвергается атероматозному распаду, вследствие чего бляшка достигает в некоторых случаях наружной оболочки сосудов. Атероматоз — начало осложненных поражений (см. далее). При прогрессировании атероматоза в связи с разрушением новообразованных сосудов происходит кровоизлияние в толщу бляшки (интрамуральная гематома). В случае разрушения покрышки бляшки образуется атероматозная язва. Дефект внутренней оболочки сосуда часто прикрывается тромботическими массами.
Атерокалъциноз — завершающая стадия морфогенеза атеросклероза, хотя отложение извести начинается уже в стадии атероматоза и даже липосклероза.
Клинико-морфологические формы.В зависимости от преимущественной локализации в том или ином сосудистом бассейне, осложнений и исходов, к которым он ведет, выделяют следующие клинико-морфологические формы атеросклероза:
▲ атеросклероз аорты;
▲ атеросклероз венечных артерий сердца (сердечная форма, ишемическая болезнь сердца);
▲ атеросклероз артерий головного мозга (мозговая форма, цереброваскулярные заболевания);
▲ атеросклероз артерий почек (почечная форма);
▲ атеросклероз артерий кишечника (кишечная форма);
▲ атеросклероз артерий нижних конечностей.
При каждой из названных форм могут наблюдаться двоякие изменения. Медленное сужение питающей артерии атеросклеротической бляшкой приводит к хронической недостаточности кровоснабжения и ишемическим изменениям — дистрофии и атрофии паренхимы, диффузному или мелкоочаговому склерозу стромы. Острая окклюзия питающей артерии, обычно обусловленная осложненными поражениями (кровоизлияние в бляшку, тромбоз), приводит к острой недостаточности кровоснабжения и развитию некроза — инфаркта, гангрены. Кроме того, в ряде случаев глубокие атероматозные язвы могут привести к развитию аневризмы, т.е. выбуханию стенки артерии в участке поражения с последующим ее разрывом и кровоизлиянием.
1. Атеросклероз аорты — наиболее частая форма. Более резко он выражен в брюшном отделе и характеризуется обычно осложненными поражениями и кальцинозом. В связи с этим чаще всего сопровождается тромбозом, тромбоэмболией и эмболией атероматозными массами с развитием инфарктов и гангрены (кишечника, нижних конечностей). Нередко развивается аневризма аорты, которая может быть цилиндрической, мешковидной или грыжевидной. Стенку аневризмы в одних случаях образует аорта (истинная аневризма), в других — прилегающие к ней органы и гематома (ложная аневризма). Если кровь отслаивает среднюю оболочку от внутренней или наружной, что ведет к образованию покрытого эндотелием канала, то говорят о расслаивающей аневризме. Образование аневризмы чревато ее разрывом и кровотечением (чаще с образованием забрюшинной гематомы). Длительно существующая аневризма аорты приводит к атрофии окружающих тканей (грудины, тел позвонков), сдавлению мочеточников, артерий (чаще позвоночных ветвей, снабжающих спинной мозг). Атеросклероз дуги аорты может лежать в основе синдрома дуги аорты, а атеросклероз бифуркации аорты с ее тромбозом — вести к развитию синдрома Лериша, имеющего характерную симптоматику.
2. Атеросклероз венечных артерий сердца лежит в основе ишемической болезни сердца, морфологическим выражением которой являются очаговая ишемическая дистрофия, инфаркт миокарда, крупноочаговый (постинфарктный) и диффузный мелкоочаговый кардиосклероз.
3. Атеросклероз артерий головного мозга является основой цереброваскулярных заболеваний, наиболее характерными проявлениями которых являются ишемический и геморрагический инфаркт головного мозга (инсульт). Длительная ишемия коры большого мозга вследствие стенозирующего атеросклероза приводит к атрофии коры большого мозга, развитию атеросклеро-тического слабоумия.
4. При атеросклерозе почечных артерий в почках либо образуются клиновидные участки атрофии паренхимы с коллапсом и склерозом стромы, либо развиваются инфаркты с последующим формированием втянутых рубцов. Возникает крупнобугристая атеросклеротическая сморщенная почка (атеросклеротический нефросклероз). В результате ишемии почечной ткани при стено-зирующем атеросклерозе возникает симптоматическая (почечная) гипертензия.
5. Атеросклероз артерий кишечника, осложненный тромбозом, ведет к гангрене кишки. Стенозирующий атеросклероз ме-зентериальных артерий может привести к развитию ишемиче-ского колита, при котором чаще поражаются селезеночный угол и ректосигмоидные отделы толстой кишки.
6. При атеросклерозе артерий конечностей чаще поражаются бедренные артерии. Стенозирующий атеросклероз при недостаточности коллатерального кровообращения приводит к атрофии мышц и характерному симптому перемежающейся хромоты (боли, возникающие в ногах при ходьбе). Если атеросклероз осложняется тромбозом, то развивается атеросклеротическая гангрена конечности.