Лабораторная работа 3
Определение содержания углерода и водорода
методом окисления нефтепродукта
Цель работы: определить содержание углерода и водорода в нефтепродукте.
Оборудование: электропечь (или газовая горелка), кварцевая трубка, кварцевый стаканчик, трубки из молибденового стекла, капилляр, шпатель металлический, пинцет, газометр, прибор для определения элементного состава нефти (рис. 5.4).
Реактивы: кислород; серебро в виде ленты, стружки, проволоки или сетки; перхлорат магния безводный; аскарит; сахароза; янтарная кислота.
Элементный анализ на содержание углерода и водорода в нефти и нефтепродукте основан на окислении углеводородных соединений до окида углерода (IV) и воды. Сущность метода заключается в окислении кислородом продуктов термического разложения пробы нефти (нефтепродукта) с последующим поглощением образовавшихся Н2О и СО2 перхлоратом магния (техническое название ангидрон) и аскаритом (NaOH, нанесенный на асбест).
Для проведения анализа используют электропечь, обеспечивающую температуру нагрева до 950 оС. Навеску нефти (нефтепродукта) сжигают в катализаторной трубке (рис. 5.4), в которую помещают также серебро в виде ленты, стружки, проволоки или сетки. Наличие серебра необходимо для улавливания оксидов серы и галогенов, образующихся при сжигании нефтепродуктов и искажающих результаты анализа. В трубку для сжигания подают кислород из газометра через систему очистки. Для очистки образующихся при сгорании нефтепродукта газов в качестве поглотительных устройств используют трубки из молибденового стекла. Трубку для поглощения паров воды наполняют безводным перхлоратом магния, для поглощения оксида
углерода (IV) СО2 – на 3/4 объема аскаритом и на 3/4 – перхлоратом магния. Перхлорат магния обладает высокой гигроскопичностью, поэтому наполнять трубки следует как можно быстрее.
Порядок выполнения работы
1. Перед проведением анализа выполняют следующие действия.
1.1. Собирают установку для элементного анализа в соответствии со схемой, приведенной на рис. 5.4. Собранная установка должна быть герметичной (герметичность проверяют перед каждым анализом).
1.2. Перед началом работы при нагретых электропечах катализаторную трубку прокаливают в токе кислорода («холостой» опыт для определения привеса поглотительных трубок), после чего подсоединяют поглотительные трубки и пропускают через них кислород из газометра со скоростью 35 – 50 см3/мин в течение 15 – 20 мин. Затем поглотительные трубки отсоединяют, охлаждают на воздухе в течение 10 мин и после этого взвешивают с точностью до 0,01 мг. При правильно собранной аппаратуре привес поглотительных трубок не должен превышать 0,02 – 0,06 мг.
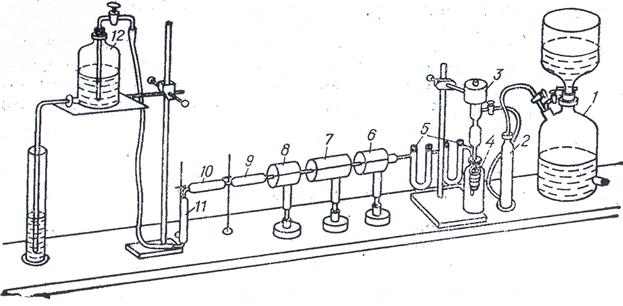
Рис. 5.4. Прибор для определения элементного состава нефти (нефтепродукта). 1 – газометр с кислородом; 2 – осушительная склянка; 3 – катализаторная трубка; 4 – сосуд для охлаждения змеевика катализаторной трубки; 5 – поглотительные трубки; 6-8 – электропечи; 9 – трубка
для поглощения воды; 10 – трубка для поглощения диоксида углерода;
11 – заключительная трубка; 12 – склянка Мариотта
1.3. Для оценки поглотительной способности аскарита и перхлората магния проводят анализ стандартного вещества, в качестве которого берут сахарозу или янтарную кислоту.
2. Выполнение анализа.
2.1. После проведения подготовительных работ берут навеску анализируемой нефти или нефтепродукта (5 – 12 мг). Необходимую навеску жидких нефтепродуктов берут при помощи капилляра, а твердых – металлическим шпателем. Навеску помещают на дно кварцевого стаканчика. Затем через кварцевую трубку для сожжения пропускают кислород со скоростью 35 – 50 см3/мин. Стаканчик с навеской нефти (нефтепродукта) быстро вносят в катализаторную трубку для сожжения и присоединяют поглотительные трубки. После сожжения анализируемой нефти (нефтепродукта) катализаторную трубку прокаливают по всей длине.
По окончании анализа поглотительные трубки отсоединяют от установки, охлаждают на воздухе в течение 10 мин и взвешивают с точностью до 0,01 мг.
Массовую долю углерода и водорода в анализируемой пробе (в %) вычисляют по формулам:
С = В∙KС ∙ 100 / а,
Н = Б∙KН ∙ 100 / а,
где В – масса СО2, мг;  KС = (атомная масса углерода / молярная масса СО2) =
KС = (атомная масса углерода / молярная масса СО2) =
=0,2729; а – масса навески анализируемой нефти (нефтепродукта), мг; Б –масса Н2О, мг; KН = (молярная масса Н2 / молярная масса Н2О) = 0,1119.
Лабораторная работа 4
Определение содержания азота в нефтепродукте методом Кьельдаля
Цель работы: определить содержание азота в нефтепродукте.
Оборудование: плитка электрическая с гнездами, обеспечивающая температуру нагрева до 500 °С; колбы Кьельдаля из стекла пирекс вместимостью 100 мл; насос водоструйный; колба-парообразователь вместимостью
2 000 – 3 000 мл; холодильник Либиха; кварцевая лодочка; конденсатор
из стекла пирекс длиной 300 мм, диаметром 50 – 60 мм; колба для отгонки аммиака из стекла пирекс вместимостью 150 мл.
Реактивы: серная кислота плотностью 1,84 г/см3, гидроксид натрия, растворы 30 %-й и 0,01 М; глюкоза; сульфат меди; сульфат калия; селен металлический; перманганат калия, 0,1 М раствор; бийодат калия; индикатор Таширо (раствор 25 мг метилового красного и 83 мг метиленового синего
в 100 мл этанола).
Сущность метода Кьельдаля для анализа нефти заключается в разложении нефти (нефтепродукта) концентрированной серной кислотой плотностью 1,84 г/см3 с использованием в качестве катализатора селена в присутствии сульфата меди, сульфата калия и глюкозы и последующем окислении продуктов разложения 0,1 Мраствором перманганата калия. При этом азотсодержащие соединения превращаются в сульфат аммония, который под действием на него щелочи разлагается с выделением аммиака:
катализатор
Нефть (нефтепродукт) ® (NH4)2SО4;
(NH4)2SО4 + 2NaOH ® 2NH3 + Na2SО4 + 2Н2О
Далее аммиак количественно перегоняют с водяным паром в титрованный раствор бийодата калия
NH3 + KIO3 ∙ HIO3 ® KIO3 + NH4IO3
Порядок выполнения работы
Навеску нефти (нефтепродукта), взятую с точностью до 0,0002 г, помещают в кварцевую лодочку. Масса навески анализируемой нефти (нефтепродукта) зависит от предполагаемого содержания в ней азота:
Массовая доля азота, % до 0,5 0,5 – 1,0 1,0 – 2,0 > 2,0
Масса навески нефти
(нефтепродукта), мг 100 – 150 50 – 100 30 – 50 < 30
Кварцевую лодочку с анализируемой нефтью (нефтепродуктом) помещают в колбу Кьельдаля и добавляют в нее следующие реагенты:
H2SО4 3 мл;
глюкозу 200 мг;
сульфат меди 40 мг;
сульфат калия 200 мг;
селен металлический 40 мг.
Собирают установку в соответствии со схемой, приведенной на рис. 5.5. Данная установка позволяет выполнять параллельно три анализа.
Колбы Кьельдаля с навеской нефти (нефтепродукта) и смесью реагентов ставят на закрытую электроплитку с гнездами и нагревают до полного разложения анализируемых проб, на что указывает обесцвечивание смеси в колбах. Разложение проводят при температуре 400…420 °С. Выделяющийся при разложении газ (ядовитый селеноводород) отводится из колб Кьельдаля через газоотводную трубку,соединенную с водоструйным насосом. По окончании процесса разложения колбы Кьельдаля охлаждают и в каждую колбу приливают в два приема по 2 мл окислителя – 0,1 Мраствора перманганата калия. После добавления каждой порции окислителя реакционную смесь нагревают до появления светло-зеленой окраски. После добавления каждой новой порции окислителя колбы следует ставить на охлажденную плитку!
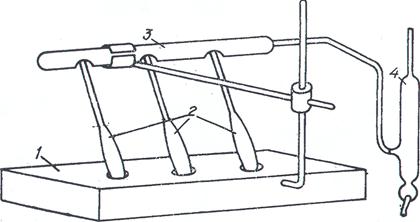
Рис. 5.5. Прибор для разложения навески нефти (нефтепродукта)
при определении азота по методу Кьельдаля: 1 – электроплитка с гнездами; 2 – колбы Кьельдаля; 3 – газоотводная трубка; 4 – водоструйный насос
Далее собирают прибор для отгонки аммиака в соответствии со схемой, приведенной на рис. 5.6, и делают предварительно холостой опыт. Перед отгонкой аммиака прибор пропаривают паром в течение 5 – 10 мин. Конденсат, собравшийся в колбе 2, титруют 0,01 М раствором NаОН в присутствии индикатора Таширо.
Анализ содержимого каждой колбы Кьельдаля проводят следующим образом. Для отгонки аммиака содержимое колбы Кьельдаля количественно переносят в отгонную колбу (рис. 5.6) и добавляют в нее 10 – 15 мл 30 %-го раствора гидроксида натрия. Из отгонной колбы аммиак отгоняют с водяным паром в течение 7 – 10 мин в приемную колбу,содержащую 9 мл бийодата калия.

Рис. 5.6. Прибор для отгонки аммиака при определении азота
по методу Кьельдаля: 1 – штатив; 2 – колба-приемник; 3 – холодильник;
4 – отгонная колба; 5 – конденсатор; 6 – стакан; 7 – воронка;
8 – парообразователь; 9 – газовая горелка (электроплитка)
Полученный в приемной колбе раствор кипятят в течение 1 мин для удаления СО2 и в горячем состоянии титруют 0,01 М раствором гидроксида натрия в присутствии смешанного индикатора Таширо до изменения окраски раствора от фиолетовой до зеленой.
Индикатор Таширо готовят следующим образом. Берут 100 мг метилового красного, 50 мг метиленового голубого и каждую навеску растворяют
в 50 мл этилового спирта; затем оба раствора сливают вместе.
Массовую долю азота N (в %) рассчитывают по формуле
N = (а – б) K × 0,14 × 100/С,
где а – количество 0,01 М раствора гидроксида натрия, израсходованное
на титрование в холостом опыте, мл; б – количество 0,01 М раствора гидроксида натрия, израсходованное на титрование в опыте с анализируемой нефтью (нефтепродуктом), мл; K – поправка к титру 0,01 М раствора гидроксида натрия; 0,14 – фактор пересчета, равный количеству азота (в мг), соответствующему 1 мл 0,01 М раствора гидроксида натрия; С – масса навески анализируемой нефти (нефтепродукта), мг.
Содержание азота в анализируемой нефти (нефтепродукте) вычисляют как среднее арифметическое трех параллельных определений.
Лабораторная работа 5
Определение содержания в нефтепродукте ванадил-
и никельпорфиринов
Цель работы: определить содержание порфиринов в нефтепродукте.
Оборудование: спектрофотометр; круглодонная колба вместимостью 0,75 – 1,0 л; обратный холодильник; воронка Бюхнера; колба Бунзена; водоструйный насос; бумажный фильтр («синяя лента»).
Реактивы: бензол; этанол; ацетон; петролейный эфир.
Теоретические сведения
В настоящее время в нефтях достоверно установлено наличие порфириновых комплексов, содержащих до 50 % ванадия и никеля. Причем до 80 – 90 % и более металлопорфиринов концентрируется в смолисто-асфальтеновой части нефти. Ванадилпорфиринов относительно больше содержится в тяжелых нефтях, а никельпорфиринов – в легких. Никельпорфирины обладают большей стабильностью, чем ванадилпорфирины. Металлопорфирины являются стабилизаторами водо-нефтяных эмульсий. Ванадиевые и никелевые порфириновые комплексы имеют биогенное происхождение: они образовались в результате металлообменных реакций из метаболических пигментов животного и растительного происхождения, таких как гемоглобин и хлорофилл.
По данным масс-спектрометрии ванадилпорфирины в нефтях представлены пятью гомологическими рядами (их обозначают М, М-2, М-4, М-6, М-8).

На гомологи рядов М и М-2 приходится обычно 90 – 95 % общего количества ванадилпрофиринов в нефти. Содержание ванадилпорфиринов в исследованных отечественных нефтях и природных битумах не превышает 100 мг на 100 г.
Металлопорфирины можно извлекать из нефти селективной экстракцией с использованием таких растворителей, как метанол, этанол, ацетон, ацетонитрил, пиридин, диметилформамид, уксусная кислота.
Практически все ванадил- и никельпорфирины находятся в смолах и асфальтенах нефти. При перегонке нефти металлопорфирины концентрируются в остаточных фракциях, выкипающих выше 500 °С.
Разработка эффективных способов выделения и очистки нефтяных металлопорфиринов даст возможность их практического использования. Например, ванадил- и никельпорфирины нефтей нашли применение при синтезе лекарственных препаратов и красителей. Аналоги металлопорфиринов – синтетические металлопорфирины (например, фталоцианины кобальта, меди, железа) являются эффективными катализаторами химических процессов.
Сущность метода определения содержания ванадил- и никельпорфиринов заключается в получении их этанольного и ацетонового экстрактов, в которых спектрофотометрическим методом определяют концентрации металлопорфиринов в видимой области спектра при длинах волн 480 – 650 нм.
Порядок выполнения работы
1. Получение этанольного экстракта металлопорфиринов. В круглодонную колбу вместимостью 0,75 – 1,0 л помещают 1 ± 0,2 г обезвоженной нефти (нефтепродукта) (для маловязких нефтей и нефтепродуктов навеску можно увеличить до 2 – 4 г).
Природные битумы, высоковязкие нефти и нефтепродукты (смолы, остатки от перегонки) разбавляют бензолом в отношении (8 ¸10) : 1 к взятой навеске, а маловязкие нефтепродукты – петролейным эфиром (фракция 40…70 оC). Затем в колбу приливают 0,5 л этанола и смесь интенсивно перемешивают. Далее колбу соединяют с обратным холодильником, устанавливают на водяной бане и кипятят в течение 2 ч (см. рис. 5.2). Затем полученный раствор отстаивают при комнатной температуре в течение 12 – 24 ч, после чего его фильтруют с помощью водоструйного насоса через бумажный фильтр («синяя лента») на воронке Бюхнера. Далее из отфильтрованного раствора этанол отгоняют простой перегонкой (см. лабораторную работу 1). Выделенный экстракт (остаток от перегонки) представляет собой окрашенную маслянистую жидкость.
2. Получение ацетонового экстракта металлопорфиринов. В колбу
с нефтяным остатком после экстракции этанолом помещают бумажный фильтр, через который фильтровали этанольный раствор, и наливают 200 мл ацетона. Затем колбу соединяют с обратным холодильником, устанавливают на водяной бане и кипятят в течение 1 ч. Далее полученный раствор отстаивают в течение 3 – 4 ч, после чего его обрабатывают так же, как и в случае экстракции этанолом.
3. Спектрофотометрическое определение концентраций ванадил-
и никельпорфиринов. Для получения спектров поглощения выделенных экстрактов используют спектрофотометр с автоматической регистрацией спектра.
Экстракты металлопорфиринов переливают в мерные цилиндры: этанольный – в градуированный цилиндр на 25 мл, ацетоновый – на 50 – 100 мл. Затем их разбавляют бензолом в таком количестве, чтобы значение оптической плотности образцов находилось в интервале 0,4 – 0,8.
Далее снимают спектры полученных экстрактов в области 480 – 650 нм
и рассчитывают содержание ванадил- (C1) и никельпорфиринов (С2) (в мг
на 100 г нефти (нефтепродукта)):
С1 = 0,187 H V / A l; С2 = 0,195H V / А l,
где 0,187 и 0,195 – коэффициенты пересчета, учитывающие поглощение
среды, оптическую плотность и молярную массу, мг/мл; Н – высота максимума полосы поглощения соответственно при 575¸5 нм для ванадилпорфиринов,. при 555¸5 нм для никельпорфиринов, см; А – масса навески нефти (нефтепродукта), г; l – толщина кюветы, см (рекомендуемая толщина кюветы l = 1 см); V – объем, до которого разбавляют экстракт бензолом, см3.
Содержание ванадил- и никельпорфиринов, определенных в этанольном и ацетоновом экстрактах, суммируют.
Лабораторная работа 6
Определение содержания в нефтепродукте смол и асфальтенов
Цель работы: определить содержание смол и асфальтенов в нефтепродукте.
Оборудование: экстракционная установка, состоящая из круглодонной колбы, экстрактора и обратного холодильника (рис. 5.7); адсорбционная стеклянная колонка высотой 1 140 мм и внутренним диаметром 18 мм с резервуаром в верхней части (длина 250 мм, диаметр 52 мм); стеклянные выпаривательные чашки диаметром 90 мм; колбы Эрленмейера вместимостью 100, 250 и 500 мл; термостат воздушный на нагрев до 200 оС; водяная баня.
Реактивы: н-гептан эталонный; толуол; спирт этиловый; спирто-толуольная смесь в соотношении 1 : 1; бензин; силикагель марки АСК с зернами размером 0,2 – 0,5 мм.
Сущность метода заключается в выделении асфальтенов н-гептаном или петролейным эфиром из нефти (нефтепродукта) и последующем отделении их фильтрованием. Смолы, растворенные в фильтрате, адсорбируются на силикагеле и затем десорбируются спирто-толуольной смесью (ГОСТ 1185).
Порядок выполнения работы
1. Подготовка установки к анализу. Силикагель насыпают в фарфоровую чашку на 3/4 емкости и помещают на 6 ч в воздушный термостат при температуре 180… ± 10 °С. Затем силикагель, не охлаждая, переносят
в сухую колбу, предварительно нагретую в течение 15 мин в том же термостате. Колбу с силикагелем плотно закрывают резиновой пробкой во избежание поглощения влаги из воздуха.
В нижнюю часть чистой сухой адсорбционной колонки закладывают тампон из стеклянной ваты и небольшими порциями насыпают охлажденный до комнатной температуры силикагель (около 100 г), непрерывно уплотняя его путем постукивания колонки в вертикальном положении. Уплотнение продолжают до тех пор, пока уровень силикагеля при встряхивании колонки не перестанет понижаться. Высота слоя адсорбента должна быть на 3-4 см ниже резервуара.
Стеклянные выпаривательные чашки (или колбы Эрленмейера) сушат
в сушильном шкафу при 105…110 °С не менее 1 ч, затем охлаждают в эксикаторе в течение 30 мин и взвешивают с точностью до 0,0002 г.
Бумажные фильтры сушат в бюксах с открытой крышкой не менее 1 ч при 105…110°С, после чего охлаждают в течение 30 мин в эксикаторе и взвешивают в закрытых бюксах с точностью до 0,0002 г.
Операции высушивания чашек или колб и фильтров повторяют до получения расхождений между двумя последовательными взвешиваниями не более 0,0004 г (до постоянной массы).
2. Выполнение анализа. Пробу нефти (нефтепродукта) тщательно перемешивают в течение 5 мин. Навеску анализируемой пробы массой 3 – 10 г взвешивают в колбе экстракционного аппарата (или колбе Эрленмейера) вместимостью 500 мл с точностью до 0,01 г и разбавляют 40-кратным количеством н-гептана (в случае необходимости при осторожном нагревании) или 30-кратным количеством петролейного эфира.
Для осаждения асфальтенов из раствора нефти (нефтепродукта) в н-гептане его отстаивают в темном месте в течение 16 ч при комнатной температуре.
Для осаждения асфальтенов из раствора нефти (нефтепродукта) н-гептаном собирают экстракционную установку (рис. 5.7), ставят ее на водяную баню с температурой 50…55 °С и выдерживают при этой температуре 30 – 35 мин, поддерживая температуру добавлением горячей воды в баню (применение электроподогрева исключается в связи с пожароопасностью). По истечении 30 мин оставляют раствор в экстракторе еще на 1 ч. Установка должна быть защищена от воздействия солнечного света.

Рис. 5.7. Экстракционная установка
3. Определение асфальтенов. В коническую воронку вставляют фильтр «синяя лента». Отстоявшийся раствор н-гептана осторожно, без перемешивания, фильтруют. Затем осадок количественно переносят на тот же фильтр с использованием н-гептана и промывают растворителем до тех пор, пока он не будет стекать совершенно прозрачным и после его испарения на фильтровальной бумаге не будет оставаться масляное пятно. Если на фильтре образуется твердый трудносмываемый слой асфальтенов, его растворяют в небольшом количестве бензола и переносят в колбу, в которой далее экстрагируют бензолом асфальтены.
Для удаления из осадка асфальтенов соосажденных вместе с ними смол и парафинов сворачивают фильтр и помещают его в экстрактор. В колбу экстракционного аппарата вместимостью 100 мл наливают 50 мл н-гептана. Собирают установку (см. рис. 5.7) и помещают ее на электроплитку (или на водяную баню, нагретую до 50…55 °С). Экстрагирование смол и парафинов проводят в течение 1 ч с н-гептаном.
Укладывать фильтр и собирать экстракционную установку необходимо по возможности быстро, так как в контакте с кислородом воздуха асфальтены переходят в труднорастворимое состояние.
Перед началом экстрагирования нагрев содержимого колбы проводят таким образом, чтобы фильтр с осадком в экстракторе полностью заполнился растворителем, и после этого ведут экстрагирование со скоростью 2-4 капли экстракта в секунду.
При использовании в качестве растворителя н-гептана проводят дополнительное экстрагирование этанолом для более полного удаления из асфальтенов высокоплавких церезинов. Для этого по окончании экстрагирования
н-гептаном колбу, в которой он находился, заменяют другой колбой вместимостью 100 мл, куда в качестве растворителя наливают 50 мл этилового спирта, и проводят экстрагирование спиртом в течение 5 – 10 мин.
По окончании экстрагирования смол и парафинов колбу с этанолом
заменяют колбой вместимостью 500 мл, в которой происходило осаждение асфальтенов (см. п. 2), предварительно налив в нее 50 – 100 мл толуола. Экстракционный аппарат помещают на электрическую плитку и ведут экстрагирование со скоростью 2-4 капли фильтрата в секунду до тех пор, пока не растворятся все асфальтены и толуол не будет стекать в колбу бесцветным.
Толуольный экстракт переносят количественно в стеклянную чашку или колбу Эрленмейера и выпаривают толуол из чашки или отгоняют из колбы Эрленмейера методом простой перегонки на водяной бане (см. рис. 5.1).
Полученные асфальтены доводят до постоянной массы нагреванием при 105 °С. Чистые асфальтены – хрупкие и блестящие вещества черно-коричневого цвета. Матовый и мазеобразный вид асфальтенов указывает на присутствие в них масел, парафинов и необходимость повторного переосаждения.
4. Определение в пробе карбенов и карбоидов. После растворения асфальтенов в толуоле фильтр с нерастворившимся остатком вынимают из экстрактора и доводят до постоянной массы. Разность между массой фильтра
с остатком и массой чистого фильтра представляет собой сумму механических примесей, карбенов и карбоидов в исследуемой пробе.
5. Определение в пробе силикагелевых смол. Фильтрат, полученный после фильтрования и промывки асфальтенов в п. 3, помещают в колбу Эрленмейера вместимостью 250 мл и выпаривают до получения 50 – 70 мл остатка. Таким образом получают концентрат фильтрата. К остатку в колбе после удаления н-гептана добавляют экстракт этилового спирта после удаления из асфальтенов высокоплавких церезинов и отгоняют растворитель до его полного удаления, а затем остаток растворяют в 30 – 50 мл бензина.
В адсорбционную колонку, заполненную силикагелем, через воронку наливают 200 мл бензина-растворителя. Когда бензин полностью впитается силикагелем, кран закрывают и в колонку загружают концентрат фильтрата. Колбу из-под концентрата промывают небольшим количеством растворителя, который также заливают в колонку. Затем в колонку заливают еще около 100 мл бензина, чтобы поверхность силикагеля была покрыта растворителем. Колонку сверху закрывают ватой и оставляют на 1 – 2 ч. Затем открывают пробку и краном регулируют скорость прохождения раствора, которая должна составлять 5 мл/мин.
Когда уровень бензина в колонке дойдет до поверхности силикагеля, в нее наливают отдельными порциями по 100 мл растворитель (смесь бензина и толуола, взятых в соотношении 6 : 1), пока из колонки не будет стекать чистый растворитель. Чистому растворителю дают полностью стечь из колонки. На промывку расходуют 500 – 600 мл растворителя.
Затем колбу с растворителем убирают, устанавливают чистую сухую колбу вместимостью 500 мл и собирают в нее растворитель после десорбции смол из силикагеля.
Для десорбции анализируемых смол из силикагеля в колонку небольшими порциями приливают 400 мл спирто-толуольной смеси. Десорбцию проводят до полного осветления растворителя, стекающего из колонки.
Из полученного после десорбции раствора на водяной бане выпаривают спирто-толуольную смесь. Выпаривание проводят в вытяжном шкафу в стеклянных чашках, доведенных до постоянной массы. Полученный сухой остаток высушивают в сушильном шкафу при 105…110 °С и доводят до постоянной массы. За постоянную массу принимают массу сухого остатка при расхождении между двумя последовательными взвешиваниями не более 0,001 г.
Массовую долю асфальтенов А (в %) вычисляют по формуле
A = (m1 / m) 100,
где т – масса навески нефти (нефтепродукта), г; т1– масса асфальтенов, г.
Суммарную массовую долю карбенов и карбоидов Х1(в %) определяют по формуле
X1 = (т2 / т) 100– С,
где т – масса навески нефти (нефтепродукта), г; т2– разность между массой фильтра с нерастворившимся остатком и массой чистого фильтра, г; С – содержание механических.примесей, %.
Массовую долю силикагелевых смол Х2(в%) находят по формуле
Х2 = (т3 / т) 100,
где т – масса навески нефти (нефтепродукта), г; т3 – масса смол, г.
Лабораторная работа 7
Определение кислотного числа нефти (нефтепродукта)
Цель работы: определить кислотное число нефти (нефтепродукта).
Оборудование: колба коническая вместимостью 250 см3, обратный холодильник, электроплитка, хлоркальциевая трубка, бюретка.
Реактивы: индикаторбромтимоловый синий, гидроксид калия (0,05 М спиртовый раствор), нефть (нефтепродукт).
Теоретические сведения
Кислотность нефти обусловлена содержанием в ней алифатических
и циклических карбоновых кислот, фенолов и других соединений кислотного характера. Все эти вещества, выделяемые из нефти растворами щелочей, называют нефтяными кислотами.
Содержание кислорода в элементном составе нефти находится в прямой зависимости от содержания нефтяных кислот. Эти кислоты образуются при биохимическом окислении нефтей. Кислородсодержащие соединения также могут образовываться в некоторых процессах добычи и переработки нефти (например, при окислении кислородом воздуха нефти при ее добыче методом внутрипластового горения и остатков нефти при производстве битумов).
Содержание нефтяных кислот в нефтях может составлять от следов до 1 % (по массе) и более. Наибольшее их количество содержится в нефтях нафтенового типа, наименьшее – в высокопарафинистых.
Нафтеновые кислоты различных нефтей представляют собой монокарбоновые кислоты ряда циклопентана и циклогексан, причем преобладают кислоты ряда циклопентана. Наряду с моноциклическими нафтеновыми кислотами в нефтях содержатся би- и полициклические кислоты. Карбоксильная группа почти во всех известных нафтеновых кислотах отделена от циклической части молекулы одной или несколькими метиленовыми группами.
В нефтях обнаружено более 20 алифатических карбоновых кислот (муравьиная, уксусная, пропионовая, масляная, капроновая, энантовая, пальмитиновая, стеариновая, миристиновая, арахиновая и др.). Муравьиная и уксусная кислоты были впервые обнаружены в кавказских нефтях выдающимся русским химиком В. В. Марковниковым еще в 1883 г. В самотлорской нефти карбоновые кислоты представлены в основном алифатическими кислотами
с числом углеродных атомов от 8 до 19, при этом преобладают кислоты
с четным числом атомов углерода, а пальмитиновая кислота (С1бН32О2) содержится в аномально большом количестве.
В нефтях обнаружены фенол и его соединения: алкилфенолы (крезолы, ксиленолы), циклоалкилфенолы, дициклоалкилфенолы, нафтолы. Трудность выделения и исследования фенолов связана с их незначительным содержанием в нефтях (менее 0,1 %) и легкой осмоляемостью. Например, в самотлорской нефти алкилфенолов состава C8Hl0O – C14H22О содержится 0,0083 %, циклоалкилфенолов – 0,0010 %, дициклоалкилфенолов – 0,0003 %, нафтолов – 0,0014 %.
Нефтяные кислоты влияют на такие процессы нефтедобычи и нефтепереработки, как нефтевытеснение, эмульгирование, отложение парафинов
и солей, коррозия оборудования. Большой коррозионной активностью обладают низкомолекулярные алифатические кислоты. Из-за коррозионной активности нефтяных кислот их содержание в моторных топливах и смазочных маслах должно быть минимальным. Кислотность автомобильных бензинов не должна превышать 3 мг КОН/100 мл бензина, авиационных – 1, реактивных топлив – 0,7, дизельных топлив – 5 мг КОН/100 мл бензина.
Соли нафтеновых кислот широко применяют в народном хозяйстве: в производстве моющих средств – соли щелочных металлов; в производстве смазочных масел – нафтенаты алюминия, кальция, свинца, бария, цинка, натрия, олова, кобальта, никеля; в лакокрасочной промышленности – нафтенаты свинца, кобальта, марганца, алюминия, кальция, магния, железа; в качестве фунгицидов и инсектицидов – нафтенаты меди, цинка, ртути; в качестве катализатора процесса окисления парафинов – нафтенат марганца; в качестве компонента зажигательных веществ (напалм) – нафтенат алюминия; нафтенат хрома является хорошим клеящим веществом. Нафтеновые кислоты и их соли в больших количествах используют при производстве лаков.
В промышленных условиях нефтяные кислоты извлекают из светлых нефтепродуктов водными растворами гидроксида натрия при 35…70 °С, а из масляных дистиллятов – при 130…160 °С и под давлением.
Содержание кислых соединений в нефти (нефтепродукте) характеризуется кислотным числом – это число миллиграммов гидроксида калия КОН, которое необходимо для нейтрализации всех кислых веществ, содержащихся в 1 г или в 100 мл нефти (нефтепродукта).
В лабораторных условиях нефтяные кислоты из нефти (нефтепродукта) выделяют различными способами спирто-щелочной обработки. Например, методом «горячего» омыления, который заключается в следующем. Разбавленную бензолом нефть смешивают с 1 М спиртовым раствором гидроксида калия и кипятят в течение 40 мин. Полученную смесь разбавляют водой, после отстаивания отделяют водный слой, который упаривают и подкисляют
10 %-м раствором серной кислоты при 0 °С до рН 5-6. Затем нефтяные кислоты экстрагируют диэтиловым эфиром или хлороформом. При «холодном» методе омыления нефть перемешивают при 20 °С в течение 30 мин с 1 М раствором гидроксида калия в 70 %-м этаноле. Затем спирто-щелочной экстракт отделяют от нефти и упаривают в вакууме при 40 °С. Экстракцию нефтяных кислот после подкисления проводят диэтиловым эфиром.
Порядок выполнения работы
(Работа производится в вытяжном шкафу!)
1. Готовят раствор индикатора: 0,5 г сухого индикатора бромтимолового синего растворяют в 80 мл воды и доводят объем раствора до 100 мл этанолом.
2. В коническую колбу вместимостью 250 мл наливают 25 мл 85 %-го этилового спирта и кипятят с обратным холодильником (водяным или воздушным) в течение 5 мин (рис. 5.8). В прокипяченный спирт добавляют 8 – 10 капель (0,25 мл) индикатора бромтимолового синего (0,5 %-й p-p) и горячий раствор нейтрализуют при непрерывном перемешивании 0,05 М спиртовым раствором гидроксида калия до первого изменения желтой окраски в зеленую (или сине-фиолетовую).

Рис. 5.8. Схема установки для определения кислотного числа нефте-продукта: 1 – электрическая плитка, 2 – колба с анализируемой смесью,
3 – обратный холодильник, 4 – хлоркальциевая трубка
3. Берут навеску нефти (нефтепродукта) массой 10 г. В колбу с навеской испытуемой нефти (нефтепродукта) приливают нейтрализованный горячий спирт и кипятят смесь в течение 5 мин с обратным холодильником при постоянном перемешивании (рис. 5.8). Если содержимое колбы после кипячения сохраняет зеленую окраску, анализ прекращают и считают, что кислотность испытуемой пробы отсутствует.
При изменении окраски до желтого цвета горячую смесь титруют спиртовым раствором гидроксида калия при непрерывном интенсивном перемешивании до изменения желтой окраски спиртового слоя или смеси в зеленую. Окраска должна быть устойчивой без перемешивания в течение 30 с. Титрование горячего раствора проводят быстро, чтобы избежать влияния диоксида углерода воздуха на содержимое колбы.
Кислотное число (КЧ) (в мг КОН/г) вычисляют по формуле
KЧ = V T / m,
где V – объем 0,05 М спиртового раствора гидроксида калия, израсходованный на титрование, мл; Т – титр 0,05 М спиртового раствора гидроксида
калия, мг/мл; m – масса навески нефти, г.
Результаты анализа округляют до второго десятичного знака. За результат анализа принимают среднее арифметическое трех параллельных определений. Расхождение между значениями параллельных определений не должно превышать следующих значений:
Кислотное число, мг КОН/г до 0,5 0,5 – 1,0 > 1,0
Ошибка измерения, % 0,06 0,10 0,20