Проводники, полупроводники, диэлектрики.
По электрическим свойствам материалы делятся на диэлектрики, проводники, полупроводники и сверхпроводники. Они отличаются друг от друга по величине удельного сопротивления, характеру изменения его в зависимости от температуры и механизму проводимости.
Диэлектрики. В отличие от металлов, кристаллы простых веществ, образованных неметаллами, обычно не обладают заметной электронной проводимостью; они представляют собой диэлектрики. Хотя в этом случае тоже возможно образование энергетических зон, но здесь зона проводимости отдельна от валентной зоны запрещённой зоной, т.е. значительным энергическим промежутком. Энергия слабого электрического поля оказывается недостаточной для преодоления этого промежутка, и электроны не переходят из валентной зоны в зону проводимости. Таким образом, в изоляторах электроны не могут свободно перемещаться по кристаллу и служить переносчиками электрического тока. Вид химической связи в основном ионный или ковалентный. Свободные носители заряда отсутствуют. Между валентной зоной и зоной проводимостью находиться широкая запрещённая зона. Основные диэлектрики: соли, оксиды, стекло, полиэтилен, резина и др. Диэлектрики поляризуются в электрическом поле. Под действием электростатического поля положение и величина эффективных зарядов атомов диэлектрика изменяются, при этом внутри диэлектрика возникает собственное энергетическое поле, направленное противоположно к внешнему. Имеются также диэлектрики с высокой диэлектрической проницаемостью. Это обусловлено наличием самопроизвольно поляризующихся областей.
Полупроводники. При нагревании они близки к проводникам, а при охлаждении к диэлектрикам. Из простых это P, I, B, Se. Также многие бинарные соединения ZnO, FeO. Зависимость электрических свойств от температуры и освещённости объясняется электронным строением их кристаллов. Здесь, как и у изоляторов, валентная зона отделена от зоны проводимости запрещённой зоной. Однако ширина запрещённой зоны, в случае полупроводников не велика. Поэтому при повышении температуры или освещённости электроны, занимающие верхние уровни валентной зоны, могут переходить в зону проводимости и участвовать в переносе электрического тока. С повышением освещённости или температуры число электронов, переходящих в зону проводимости, возрастает, в соответствии с этим увеличивается и электрическая проводимость полупроводника. В полупроводниках с ковалентной химической связью, появление электронов в зоне проводимости одновременно создаёт его вакансию в валентной зоне. Данная вакансия на конкретной молекулярной орбитали может заполняться электронами других занятых ближайших МО. Такой переход электронов внутри валентной зоне как бы создаёт движение вакансий с одного МО на другую МО. Поэтому электрический ток в полупроводнике определяется движением электронов в зоне проводимости и в валентной зоне. Полупроводники применяются в радиоэлектронике.
Проводники. С повышением температуры они увеличивают свою проводимость. Носителями заряда служат электроны. Валентная зона и зона проводимости электронной структуры пересекаются. Это позволяет электронам из валентной зоны при небольшом возбуждении переходить на молекулярные орбитали зоны проводимости, а это значит, что электрон с другой вероятностью появляется в той или иной точке компактного металла. Проводники используются для передачи электроэнергии. Среди проводников выделяются: металлы (Al, Cu, Fe) и сплавы высокой проводимостью (латунный, бронзовый, алюминиевый).
Особенности хим.связи в металлическом и атомном кристалле. По прочности металлическая решётка находиться между атомными и молекулярными металлическими решётками. Это связано с тем, что металлической связи характерны черты ковалентной связи и отдельные черты дальнодействующей связи. Металлическая решётка бывает и малопрочная (ртуть). Упрощенно металлическая решетка представляется в виде положительно заряженных ионов, располагающихся в узлах ее, и электронов, двигающихся между ними. Атомы металлов, с характерным для них дефицитом валентных электронов, должны иметь как можно больше соседних атомов, чтобы этот дефицит компенсировать за счет электронов соседей. Кристаллическое состояние вещества. Существуют вещества, в кристаллах которых значительную роль играют несколько видов взаимодействия между частицами. Так, в графите атомы углерода связаны друг с другом в одних направлениях ковалентнымй связями локализованного и делокализованного характера, а в других — межмолекулярной связью. Поэтому решетку графита можно рассматривать и как атомную, и как металлическую, и как молекулярную. В узлах атомных решеток находятся атомы; они связаны друг с другом ковалентной связью. Веществ, обладающих атомными решетками, сравнительно мало. К ним принадлежат алмаз, кремний и некоторые неорганические соединения. Эти вещества характеризуются высокой твердостью (алмаз). Они тугоплавкие и практически не растворимы. Такие из свойст обусловлены прочностью ковалентной связи.
Чистые i - полупроводники практически не используют. В них специально вводят атомы других элементов (примеси) трехвалентных (алюминий, галлий, индий, бор) или пятивалентных (мышьяк, фосфор, сурьма) элементов или их соединений. При этом на 107…108 атомов i - полупроводника вводят один атом примеси. Атомы пятивалентной примеси называются донорами: они увеличивают число свободных электронов. Каждый атом такой примеси добавляет один лишний электрон. При этом лишних дырок не образуется. Примесный атом в структуре полупроводника превращается в неподвижный положительно заряженный ион. Проводимость полупроводника теперь будет определяться в основном числом свободных электронов примеси. В целом такой тип проводимости называют проводимостью n–типа, а сам полупроводник – полупроводником n–типа.
При введении трехвалентной примеси одна из валентных связей полупроводника оказывается незаполненной, что эквивалентно образованию дырки и неподвижного отрицательно заряженного иона примеси. Таким образом, в этом случае увеличивается концентрация дырок. Примеси такого типа называются акцепторами, а проводимость, обусловленная введением акцепторной примеси, называют проводимостью р–типа. Полупроводник данного вида называют полупроводником р–типа.
Преобладающие носители заряда в полупроводнике называются основными. Так в полупроводнике n–типа основными носителями являются электроны, а неосновными – дырки, а в полупроводнике р–типа основными носителями являются дырки, а неосновными – электроны. Как видим, в отличие от проводимости проводников, в которых ток обусловлен направленным движением только электронов, в полупроводниках ток может быть обусловлен двумя типами носителей – электронами и дырками.
22.Энтропия и ее изменение при химических реакциях.
Мера вероятности состояния системы принятых называть - Энтропия(S)- величина пропорциональная log числа равновероятных микросостояний которыми может быть реализовано данное макросостояние(Т,Р,V). Энтропия имеет размерность энергии, деленной на температуру. Выражают в [ДЖ/моль*кельвин]. S возрастает при переходе в-ва из кристаллического состояния в жидкое и из жидкого в газообразное, при растворении кристалло, при расширении шазов, при хим.взаимодействиях, приводящих к увеличению числа частиц, и прежде всего частиц в газообразном состоянии. Напротив, все процессы в результате которых упорядоченность системы возрастает(конденсация, полимеризация, сжатие, уменьшения числа частиц), сопровождаются уменьшением энтропии.
23.Энергия Гиббса. Условия самопроизвольного протекания химических процессов.
Энергия Гиббса(G)- называемая также изобарно-термическим потенциалом или свободной порцией при постоянном давлении. G связана с H,S,T: G=H-TS. Если реакция осуществляется при постоянных P и Т, то ∆G=∆H-T∆S. При постоянстве температуры и давления хим.реакции могут самопроизвольно протекать только в таком направлении, при котором энергия Гиббса системы уменьшается. При низких температурах самопроизвольно могут протекать экзотермические реакции, а при высоких – рекции сопровождающиеся влечение энтропии.
| Знак изменения ф-ии | Самопроизвольное протекание | ||
| ∆Н | ∆S | ∆G | |
| - + - + | + - - + | - + +/- +/- | Возможно при любых температурах Невозможно при любых температурах Возможно при достаточно низких температурах Возможно при достаточно высоких температурах. |
24.Предмет химической кинетики. Скорость химических реакций и факторы, влияющие на нее при гомогенных и гетерогенных процессах.
Хим.кинетика- раздел химии изучающий скорости хим.реакций. Скорость хим.реакции измеряется количеством в-ва, вступившего в реакцию или образующегося в результате реакции в единицу времени в единице объема системы(для гомогенной реакции) или на единице площади поверхности раздела фаз(для гетерогенной реакции). В случае гомогенного процесса протекающего при постоянном объеме, скорость гомогенной реакции измеряется изменением концентрации какого-либо из реагирующих в-в за единицу времени. Скорость реакции зависит от природы реагирующих в-в их концентрации, температуры и от присутствия в системе катализаторов. Влияние температуры. Зависимость скорости реакции от температуры определяется константой скорости. Правило Вант-Гоффа: при повышении температуры скорость большинства химических реакций возрастает в 2-4 раза при нагревании на каждые 10 градусов. V(T2)/V(T1)=y(T2-T1)|10
25.Влияние концентрации на скорость химической реакции. Закон действующих масс.
Закон действующих масс(основной закон химической кинетики): при постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ. A+2BàAB2 u=k[A][B][B]=k[A][B]2, где k-константа скорости реакции, значение которой зависит от природы реагирующих вещ-в. При гетерогенных реакциях концентрации веществ, находящихся в твердой фазе, обычно не изменяются в ходе реакции и поэтому не включаются в уравнение закона действия масс. CaCO3(к.)àCaO(к.)+CO2(г.) u=k,т.е u=const,т.к. CaCo3 – твердо в-во.
Билет №26 Энергия Активации. Влиян. Т на V
Энергия активации- некоторая избыточная энергия (по сравнению со средней энергией молекул при данной температуре), которой должны обладать молекулы для того, чтобы их столкновение было эффективным, т.е привело к образованию нового в-ва. С ростом температуры число активных молекул быстро увеличивается, что приводит к резкому возрастанию скорости реакции. ((Уравнение Аррениуса: k=ZPe-Ea/RT, Z-число столкновений молекул в секунду в единице объема; e=2, 718..; R-универсальная газовая постоянная = 8,314 Дж/моль*К; Т- температура, К; Р- так называемый стерический множитель. ))
26.Энергия активации. Влияние температуры на скорость химической реакции.
Энергия активации- некоторая избыточная энергия (по сравнению со средней энергией молекул при данной температуре), которой должны обладать молекулы для того, чтобы их столкновение было эффективным, т.е привело к образованию нового в-ва. Катализатор – в-во, не расходующееся в результате протекания реакции, но влияющее на ее скорость. Катализ – явление изменения скорости реакции под действием таких веществ. Бывают катализаторы как ускоряющие протекание реакции, так и замедляющие ее. В первом случае катализ называется положительным, а во втором - отрицательным.
Ингибиторы- катализаторы, уменьшающие скорость реакции.
27.Энергия активации. Представление о катализе. Катализаторы и ингибиторы
Энергия активации- некоторая избыточная энергия (по сравнению со средней энергией молекул при данной температуре), которой должны обладать молекулы для того, чтобы их столкновение было эффективным, т.е привело к образованию нового в-ва. Катализатор – в-во, не расходующееся в результате протекания реакции, но влияющее на ее скорость. Катализ – явление изменения скорости реакции под действием таких веществ. Бывают катализаторы как ускоряющие протекание реакции, так и замедляющие ее. В первом случае катализ называется положительным, а во втором - отрицательным.
Ингибиторы- катализаторы, уменьшающие скорость реакции.
28. Реакции простые и сложные. Лимитирующая стадия химического процесса.
Простые: происх. между простыми в-ми. Сложные: между сложными. Большинство химических реакций идут не в одну стадию, но и одностадийная реакция может кинетически осложняться, если она обратима. К кинетически сложным реакциям относят также последовательные(Последовательными называют реакции с промежуточными стадиями, когда продукт предыдущей стадии служит исходным веществом для последующей При k1 >> k2 все исходное вещество может превратиться в промежуточный продукт В, прежде чем начнется вторая реакция. Скорость всей реакции определяется второй стадией. При k1 << k2 концентрация промежуточного продукта мала, поскольку он не успевает накапливаться; эта стадия определяет скорость реакции в целом. Таким образом, скорость определяется самой медленной стадией (принцип лимитирующей стадии)). параллельные(Реакции, в которых исходные вещества способны образовывать разные продукты реакции или одно вещество одновременно способно реагировать с несколькими веществами, называются параллельными.), цепные(К цепным реакциям относят реакции, протекающие с образованием свободных радикалов, способных превращать реагенты в конечные продукты, поддерживая постоянство свободных радикалов или даже увеличивая их (разветвленная цепная реакция). Стадия определяющая скорость протекания реакции – есть лимитирующая стадия.
29.Константа равновесия. Возможность смещения равновесия химической реакции в прямом и обратном направлениях. Принцип Ле-Шателье.
Константа равновесия: величина с помощью которой описывается равновесие между веществами в растворе, преимущественно между газами и жидкостями. Зависит только от температуры. Равновесный закон действующих масс: отношения произведения молярных концентраций продуктов к произведению молярных концентраций реагентов взятых в степенях равных стехиометрическим коэффициентам есть величина постоянная при данной температуре =Kр [C]с[D]d/[A]a[B]b=K. Процесс изменения концентраций, вызванный нарушением равновесия, называется смещением или сдвигом равновесия. При увеличении концентрации одного из веществ равновесие смещается в сторону расхода этого вещ-ва. При увеличении давления равновесие сдвигается в сторону уменьшения числа молекул газов т.е. в сторону понижения давления. При повышении температуры равновесие смещается в сторону эндотермической реакции. Принцип Ле-Шателье: Если на систему, находящуюся в равновесии, оказать какое либо воздействие, то в результате протекающих в ней процессов равновесие сместится в таком направлении, которое уменьшит указанное воздействие.
30.Область жидкого состояния вещества. Диаграмма состояния воды.
Жидкостью называется физическое тело, обладающее двумя отличительными особенностями: незначительным изменением своего объема под действием больших внешних сил и текучестью, то есть изменением своей формы под действием даже незначительных внешних сил. Плотность жидкостей уменьшается с увеличением температуры. Исключение представляет вода в диапазоне температур от 0 до 40С, когда ее плотность увеличивается, достигая наибольшего значения при температуре 40С r = 1000 кг/м3. Диаграмма состояния воды показывает графическое изображение зависимости между величинами, характеризующими состояние системы и фазовыми превращениями в системе.
31.Способы выражения количественного состава растворов: массовая, объемная и мольная доли. Массовая, молярная и нормальная концентрации, моляльность.
Растворы – это гомогенные (однофазные) системы переменного состава, состоящие из двух или более веществ (компонентов). Раствор находящийся в равновесии с растворяющимся веществом называется насыщенным. Раствор с низким содержанием растворённых веществ- разбавленный, наоборот-концентрированный. Способы выражения: 1. Массовая доля: отношение массы растворённого вещества к массе раствора. 2. Мольная доля: отношение кол-ва растворённого вещ-ва к сумме кол-в всех веществ составляющих раствор. N=n2\(n1+n2) 3. Молярность(Cm): отношение кол-ва вещ-ва к объёму раствора. 4. Моляльность(m): отношение кол-ва вещ-ва к массе р-ля. 5. Эквивалентная или нормальная конц-ия(Сн): отношение числа эквивалентов к объёму раствора. V1\V2=N2\N1 Объёмы растворов реагирующих веществ обратно пропорциональны их нормальностям. Объемная доля растворенного в-ва представляет собой отношение объема компонента, содержащегося в системе, к общему объему системы(ф=V(растворен)/V(раствора)*100%).
32.Растворы. Тепловые эффекты при растворении.
Раствор- гомогенная система которая состоит по крайней мере из 2 веществ, одно из которых находится в избытке. Растворение похоже на хим. Реакцию но состав раствора может изменятся в широких пределах. Кроме того в свойствах растворов обнаруживаются свойства отдельных компонентов. Таким образом раствор занимает промежуточную стадию между хим. Соед. И механическими смесями. В зависимости от размера частиц подразделяются на: Истинные (частицы-молекулы, ионы <10-7) коллоидные (макромолекулы, ионные пары, атомы металлов10-710-5). Термодинамические факторы процесса растворения: Большинство веществ растворяются в жидкости с поглощением теплоты, но это не всегда так. Кол-во теплоты, поглощающейся(или выделяющейся) при растворении одного моля вещества, называется теплотой растворения этого вещества. При растворении энергия затрачивается на разрушение кристаллов растр. В-ва т.е. происходит поглощение энергии, если этого не наблюдается значит между растворителем и растр. В-ом происходит химическое взаимодействие. Такие соединения – сольваты. Растворимость твёрдых веществ повышается с увеличением Т, газов - наоборот.
33.Растворимость. Коэффициент растворимости. Селективные растворители и их использование в процессах осушки и очистки природных газов.
Растворимость-способность в-ва растворятся в том или ином растворителе. Мерой растворимости в-ва при данных условиях служит содержание его в насыщенных растворах.; равновесная концентрация растворённого в-ва в его насыщенном растворе. Если в 100г. Воды раствор. Более 10г. В-ва –хорошо растворимое. Число единиц массы безводного в-ва насыщающего при НУ 100г. Единиц массы р-ля – есть коэффициент растворимости. Температура при которой ограниченная растворимость становится неограниченной называется критической температурой растворения Селективный растворитель: Диметилформамид Основные промышленные применения
34.Представление об идеальных растворах. Влияние концентрации растворенного вещества на давление насыщенного пара раствора, его температуру кипения и кристаллизации (закон Рауля и следствия из него).
Идеальный раствор
Закон Рауля: Относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворённого вещества. (Po-P)/Po=N Второй закон Рауля–понижение температуры кипения и повышение температуры замерзания раствора прямо пропорционально молярной концентрации раствора.
35.Явление электролитической диссоциации. Степень диссоциации. Классификация электролитов по степени диссоциации. Сильные и слабые электролиты.
Явление электролитической диссоциации: при растворении соли в воде ионы, образующие данный электролит, под действием полярных молекул воды отрываются друг от друга и перераспределяются м/у молекулами растворителя. Степень диссоциации- это отношение числа молекул, распавшихся на ионы, к общему числу растворенных молекул. Чем более полярен растворитель, тем больше степень диссоциации в нем данного электролита. Электролиты, степень диссоциации которых даже в относительно концентрированных растворах велика(à1), называют сильными(почти все соли, мин.соли : H2SO4, HNO3, HI, HMnO4; основания щел и щел-зем металлов), а электролиты, степень диссоциации которых даже в разбавленных растворах мала,-слабыми(Почти все орг. кислоты, некоторые мин.кислоты: H2CO3, H2S, многие гидрооксиды металлов).
36.Электролитическая диссоциация. Факторы, влияющие на диссоциацию электролитов. Слабые электролиты. Константа диссоциации слабых электролитов.
Электролитическая диссоциация- самопроизвольный процесс распада электролита в растворе с образованием положительно и отрицательно заряженных ионов, или соответственно катионов и анионов. 1)Чем более полярен растворитель, тем больше степень диссоциации в нем данного электролита. 2)повышение температуры, как правило, увеличивает диссоциацию и при нагревании степень диссоциации возрастает. 3)при уменьшении концентрации электролита, т.е. при его разбавлении, степень дисс. увеличивается. Электролиты, степень диссоциации которых даже в разбавленных растворах мала,- слабые(Почти все орг. кислоты, некоторые мин.кислоты: H2CO3, H2S, многие гидрооксиды металлов). Константа диссоциации сл.электролитов- это отношение произведения концентраций продуктов реакции к концентрации реагентов.
37.Диссоциация слабых электролитов. Количественные характеристики процесса диссоциации: степень и константа диссоциации. Закон разбавления Освальда.
Константа диссоциации сл.электролитов- это отношение произведения концентраций продуктов реакции к концентрации реагентов. Константа диссоциации – константа равновесия отвечающая диссоциации слабого электролита. Зависит от природы электролита и растворителя, температуры но не зависит от концентрации. Характеризует способность данного в-ва распадаться на ионы. Чем больше К тем лучше в-во диссоциирует. Кислоты и основания диссоциируют ступенчато. Каждая ступень диссоциации характеризуется своей константой. Так, трехосновная ортофосфорная кислота H3PO4 диссоциирует следующим образом….Константа равновесия Kр этой реакции и есть Kд: Kp=[H+][An]/[HAn]Если выразить равновесные концентрации через концентрацию слабого электролита C и его степень диссоциации α, то получим Kд=(C*a*C*a)/C(1-a)=(C*a2)/(1-a). Это соотношение называют законом разбавления Оствальда. Для очень слабых электролитов при α << 1 это уравнение упрощается:. Kд=(C*a2)
38.Диссоциация воды. Понятие «водородный показатель». Кислые и щелочные среды. Понятие о кислотно-основных индикаторах.
Вода достаточно плохо диссоциирует. Но всё же обладает измеримой проводимостью. Вода диссоциирует на катионы водорода и анионы гидроксила. Так как степень диссоциации воды крайне мала то концентрация недиссоциированных молекул H2O в воде практически равна общей концентрации воды. Для воды и разбавленных водных растворов при неизменной температуре произведение ионов водорода и гидроксил -ионов есть величина постоянная = ионное произведение воды.=10-14 . Растворы в которых концентрации ионов водорода = концентрации ионов гидроксила называются нейтральными([H+]=10-7). Если водорода больше([H+]>10-7) то кислый, если гидроксила([H+]<10-7) то основной но произведение ВСЕГДА остаётся постоянным. Кислотность или щелочность среды выражается водородным показателем pH=-lg[H+] Методы измерения: колориметрический и потенциометрический.
| Название индикатора | Цвет индикатора в различных средах | ||
| в кислой | в нейтральной | в щелочной | |
| Метиловый оранжевый Метиловый красный Фенолофталеин Лакмус | Красный(pH<3,1 ) Красный (pH<4,2) Бесцветный (pH<8) Красный (pH<5) | Оранжевый (3,1<pH<4,4) Оранжевый (4,2<ph<6,3) Бледно-малиновый (8<pH<9,8) Фиолетовый (5<pH<8) | Желтый (ph>4,4) Желтый (pH>6,3 ) Малиновый (pH>9,8) Синий (pH>8) |
39.Диссоциация сильных электролитов. Активность ионов в растворах. Коэффициент активности. Представление о ионной силе раствора.
В водных растворах сильные электролиты полностью диссоциируют. Поэтому число ионов в них больше чем в слабых при той же концентрации. Принципиальное отличие сильных электролитов от слабых состоит в том, что равновесие диссоциации сильных электролитов полностью смещено вправо:H2SO4>H++HSO4- Каждый ион окружен оболочкой из ионов противоположного знака. В свою очередь, каждый из этих ионов сольватирован. Это окружение называется ионной атмосферой. Активность иона – его условная эффективная концентрация соответственно которой он действует в хим. реакциях. A=Ci*f где f-коэффицент активности различный для разных ионов зависит от условий и концетрации. В конц. р-рах f<1 а с разбавлением стремится к 1. В разбавленных растворах коэф. акт. данного иона зависит лишь от ионной силы и его заряда. Где ионная сила раствора есть полусумма произведений концентраций всех находящихся в растворе ионов на квадрат их заряда. L=1/2*(C1z12+C2z22+…).
40.Кислоты и основания. Основные положения теории кислот и оснований Аррениуса, Бренстеда-Лоури, Льюиса
Теория кислот и оснований Аррениуса (1887). Теория о процессах диссоциации в водных растворах; в соответствии с ней кислоты представляют собой вещества, которые в водных растворах отдают положительные ионы водорода; основания представляют собой вещества, которые в водном растворе отдают отрицательные гидроксид-ионы. Кислота —> Анион (кислотный остаток) + Н+ Основание —* Катион + ОН~ По Аррениусу между кислотой и основанием не существует функциональной связи. Только взаимодействие катионов водорода с гидроксид-ионами с образованием воды является по Аррениусу кислотно-основной реакцией: Н+ + ОН- = Н2О Теория кислот и оснований Брёнстеда-Лоури (1923) Теория о процессах в водных и неводных растворах; кислоты являются частицами, которые способны отдавать протоны (доноры протонов);основаниями являются частицы, которые способны присоединять протоны (акцепторы протонов).Кислота (равносильно) Основание + Н+ Для каждой кислоты существует сопряженное основание, которое имеет на один протон меньше (и наоборот). Каждая кислота и каждое основание, которые находятся в такой функциональной связи, являются сопряженной парой кислота/основание.Кислотная и основная функции частиц не зависят от их заряда. Кислоты: HCI, H2SO4 NH + , H30+, НSО4. Основания: NH3, ОН', СО|-, СН3СОО-, [Zn(H20)3OH]+. Кислотно-основная реакция по Брёнстеду заключается в переносе протонов между двумя сопряженными парами кислот и оснований. HCI = Н+ + CI-Н20 + Н+ = Н3О+ HCI + Н2О= Н3О+ + CI- Амфолиты представляют собой частицы, которые в зависимости от другого участника реакции функционируют как кислота или как основание. Н20, НСО3, Н2РО4, НРО|-. Кислоты, основания и амфолиты называются протолитами. Теория кислот и оснований Льюиса (1923) Теория с расширением понятия кислот и оснований на основе электронной конфигурации; в соответствии с ней кислоты являются акцепторами электронной пары (электрофильные частицы с электронными вакансиями в их внешней электронной оболочке), основания являются донорами электронной пары (нуклеофильные частицы, в которых свободной является как минимум одна электронная пара). Кислоты: BF3, AICI3, H+, Cu2+. Основания: МН3, Н2О, ОН'. Кислотно-основная реакция является взаимодействием кислоты Льюиса с основанием Льюиса с образованием аддукта Льюиса. BF3 + NH3 F3B —NH3 Кислота Льюиса Основание Льюиса Аддукт Льюиса По Льюису кислотно-основными будут и реакции комплексообразования. Cu2+ + 4NH3 = [Cu(NH3)4]2+
41.Гидролиз. Общие закономерности гидролиза солей. Факторы, влияющие на процесс гидролиза. Гидролиз солей различной природы.
Гидролиз солей – процесс взаимодействия в-ва с водой, при котором составные части в-ва взаимодействуют с составными частями воды, образуя слабодиссоциирующие ионы или молекулы. При гидролизе происходит изменение реакции среды. Гидролизу подвергаются соединения различных классов. Гидролизу будут подвергаться соли, образованные: а)сл.основание и сильн.кислотой. б)сл.кислотой и силь.основание в)сл.кислотой и сл. основанием.; Соли образованные силь.основ и силь.кисл (NaCl ,NaNO3). Гидролиз является процессом обратимым: чем слабее электролит, образующий соль, тем глубже протекает процесс гидролиза. Гидролиз протекает тем полнее, чем слабее электролит, образовавший соль, чем выше температура и чем больше разбавление раствора.
42.Количественные характеристики процесса гидролиза: степень и константа гидролиза. Напишите уравнения реакций гидролиза солей Al2(SO4)3, Na2CO3, Сr2(СO3)3, КNO3 в молекулярной и ионно-молекулярной формах.
Степень гидролиза – доля в-ва подвергшееся гидролизу. Зависит: от константы равновесия, от температуры, от концентрации соли. Константа гидролиза – это отношение произведения концентраций кислоты и основания к концентрации образовавшейся соли. 1) Al2 (SO4)3+H2O=2AlOHSO4+H2SO4 (сл. + сильн.)
2) Na2CO3+H2O=NaHCO3+NaOH (сильн. + сл.)
3) Cr2 (CO3)3+H2O=Cr(OH)3|-+H2CO3(сл. +сл.)
4)KNO3 гидролизу не подвергается (сильн. + сильн.)
43.Равновесие в гетерогенных системах. Произведение растворимости. Условия образования и растворения осадков.
К равновесным системам следует отнести также и систему труднорастворимый электролит – его насыщенный раствор. В этом случае мы имеем дело с динамическим гетерогенным равновесием осадок-его насыщенный раствор. Например CaSO4=Ca2++SO42- Константа равновесия для этого процесса будет иметь вид: K=[ Ca2+][ SO42-]\[ CaSO4] но так как конц. тверд. соли есть Const, то Ур-е имеет вид K=[ Ca2+][ SO42-] В насыщенном растворе электролита произведение концентраций его ионов есть величина постоянная при данной Т. И обозначается произведением растворимости (ПР). Характеризует количественно способность электролита растворяться. Численное значение находится исходя из растворимости. НО Произведение растворимости вычисленное без учёта коэффициентов растворимости является постоянной величиной только для малорастворимых электролитов. ПК – произведение концентраций ионов в степенях, соответствующих стехиометрическим коэффициентам для системы в неравновесном состоянии. Зная ПК и сравнив его с ПР, можно установить, растворится или выпадет осадок при данной температуре : если ПК = ПР, ΔG = 0 – система находится в состоянии равновесия (раствор насыщенный).Если ПК < ПР, ΔG < 0 – самопроизвольно протекает процесс растворения осадка. Если ПК > ПР, ΔG > 0 – возможен только обратный процесс – выпадение осадка.
44.Жесткость природных вод. Временная и постоянная жесткость. Способы устранения жесткости. Представление о системе водоподготовки.
Минералогический состав пресной воды определяет ее жесткость (устранимую и постоянную), что требует специальной обработки перед использованием в нагревательных системах для предотвращения образования накипи. Природная вода, содержащая большое количество растворенных солей кальция и магния, называется жесткой. Соли, обуславливающие жесткость воды, не являются вредными для здоровья человека. Но наличие в воде, предназначенной для питья, большого количества магния, например, не желательно, так как он ухудшает вкус воды. Выражается в (моль/литр) ранее выражалась в градусах жёсткости. Различают жесткость: общую, временную, постоянную, карбонатную и некарбонатную.Общейназывается суммарная концентрация ионов Са2+,Mg2+ и Fe2+ в воде, выраженная в мг-экв/л. Постояннойжесткостью называется часть общей жесткости воды, остающаяся после кипячения воды при атмосферном давлении в течение 1 часа. Временной жесткостью называется часть общей жесткости, удаляющаяся кипячением воды при атмосферном давлении в течение определенного времени. Она равна разности между общей и постоянной жесткостью. Способы устранения жесткости: кипячение и применение фильтров. Система водоподготовки: 1.Грубая очистка (отделение грубодисп. систем, осаждение катионов тяжёлых металлов, газообмен при помощи кислорода) 2. Тонкая очистка (фильтрование взвешенных веществ)3.адсорбция (удаление микрозагрязнений, Аренов и пахучих веществ)4.Дезинфекция (окисление микроорганизмов).
45.Окислительно-восстановительные реакции. Окисление и восстановление. Типичные окислители и восстановители.
Окислительно-восстановительные реакции – такие реакции, в результате которых изменяется степень окисленности одного из реагирующих в-в. Окислённость – неравномерность распределения электронов между атомами. Элемент атомы которого смещаются в сторону другого элемента – «+» Окислённость, Элемент, к атомам которого смещаются электроны – «-» окисленность. Окисление – отдача атомом электронов, сопровождающаяся повышением его степени окисленнности. Восстановление- присоединение атомом электронов, приводящее к понижению его степени окисленности. Вещество в состав которого входит окисляющийся элемент – восстановитель., а вещество, содержащее восстанавливающий элемент – окислитель. Типичные восстановители: активные металлы ( щелочные и щелочноземельные, Zn, Al, Fe, и др. Me0 –ne à Me +n), а также некоторые неметаллы, H2 и C (в виде угля или кокса). Тип. окислители -типичные неметаллы (F2 ,Cl2,Br2,I2, O2), галогены, выступая в кач. окислителей , приобретают степень окисленности -1 ( от F к I окислительные св-ва ослабевают).
46.Типы окислительно-восстановительных реакций. Составление уравнений окислительно-восстановительных реакций методом электронного баланса. Приведите примеры реакций каждого типа.
Межмолекулярный тип реакции. Если окислитель и восстановитель находятся в молекулах различных в-в. ( H2S+4 O3+H2S-2=S0+H2O ) Тип Диспропорционирования или самоокисления-самовосстановления. Когда окислитель и восстановитель представлены одним и тем же в-вом с одинаковой степенью окисления ( таким двойственным св-вом обладают неметаллы, кроме F2 и O2). ( Cl20+NaOHàNaCl-+NaCl+5 O3+H2O ) Внутримолекулярный тип. Если в молекуле сложного в-ва содержатся атомы, один из которых явл. окислителем , другой – восстановителем.(N -3H4N+3 O2àN20+H2O) Метод электронного баланса. Последовательность: 1. Составить схему реакции с указанием исходных и образ веществ, отметить элементы, изменяющие в результате реакции степень окисленности, найти окислитель и восстановитель. 2. Составить схемы полуреакций окисления и восстановления с указанием исходных и образующихся реально существующих в условиях реакции ионов и молекул. 3. Уравнять число атомов каждого элемента в левой и правой частях полуреакций; при этом следует помнит, что в водных растворах в реакциях могут участвовать молекулы H2O, ионы H+ или ион ОН-. 4.Уравнять суммарное число зарядов в обеих частях каждой полуреакции; для этого прибавить к левой и правой частям полуреакции необходимое число электронов. 5. Подобрать множители (основные коэффициенты) для полуреакций так, чтобы число электронов, отдаваемых при окислении, было равно числу электронов, принимаемых при восстановлении. 6. Сложить уравнения полуреакций с учетом найденных основных коэффициентов. 7. Расставить коэффициенты в уравнении реакции.
47.Возникновение скачка потенциала на границе раздела металл/электролит. Стандартный электродный потенциал. Водородный электрод. Стандартный водородный электрод.
В силу того, что у атомов поверхности металлов валентные возможности реализованы не полностью, и на границе металл- электролит возникает промежуточная фаза, состоящая из гидратированных атомов металла. [Me*nH2O]= [Me*nH2O]+ne. Рано или поздно м/у анодным и катодным процессом установится равновесие. В силу стремления металла к окислению к моменту равновесия некоторое кол-во ионов металла остается в раст-ре электролита, а избыточное количество электронов - в металле. На границе раздела возникает скачок потенциала. Скачок потенциала, который возникает на границе раздела фаз, при переходе металла из вакуума в раствор электролита, наз. электродным потенциалом металла. Это – термодинамическая характеристика системы металл-электролит. Чем более активный металл, тем он больше стремиться к окислению, тем больше будет величина скачка потенциала на границе раздела фаз, тем более отрицательный электродный потенциал. Величину электродного потенциала измеряют путем сравнения: определяют разность потенциалов м/у рассматриваемой системой металл-электролит (рабочий электрод) и другой подобной рабочей системой, потенциал которой принят за ноль. Стандартным электродом сравнения является стандарт.(нормальн ) водородный электрод – платиновая пластинка, электролитически покрытая губчатой платиной и погруженная в раствор кислоты, через который пропускается водород.. 2H++2e=H20 Потенциал металла, измеренный при стандартных условиях относительно стандартного водородного электрода сравнения наз. стандартным электродным потенциалом.
48.Ряд стандартных электродных потенциалов. Выводы из ряда стандартных электродных потенциалов.
Когда каждая реакция(окисл\восст) протекает в гальваническом элементе Или при электролизе то каждая реакция происходит на соотв. Электроде, то такие полу-реакции называются электродными процессами. Протекающей в гальваническом элементе о\в реакции соотв ЭДС этого элемента Е связанная с изменением энергии Гиббса dG=-zFE Величины каждой полу-реакции, разность которых составляет ЭДС – электродные потенциалы. E=f1-f2 Величина потенциала зависит от: 1.природы в-в 2.отношение между концентрациями 3. температуры f=f0+(2,3RT/)*lg где f0-станд электродный потенциал R-газовая постоянная Т- абсолютная температура z- число электронов F-постоянная Фарадея [Ox] [Red] – произведения концентраций веществ участв в процессе в окисленной и восстановленной формах. Стандартный электродный потенциал при концентрациях равных единице. Для построения шкалы Эл. Потенциалов нулём была взята реакция 2H++2e-=H2 Стандартный потенциал данного процесса = 0. Все электродные потенциалы выражены в водородной шкале.
49.Гальванические элементы. Химические гальванические элементы. Анод и катод. Анодный и катодный процессы. ЭДС гальванического элемента.
Устройства, в которых энергия химической окислительно-восстановительной реакции превращается в электрическую энергию, называются источниками электрической энергии, или гальваническими элементами. Всякий гальван. элемент сост. из двух электродов – металлов, погруженных в растворы электролитов; последние сообщаются друг с другом – обычно через пористую перегородку. Переход Ме из кристалла в раствор электролита в виде гидротированных ионов с оставлением эквивалентного количества е в кристалле, называется анодным процессом - процесс окисления. Обратным анодному является катодный процесс – объединение каких-либо частиц среды с электронами металла, с восстановлением частиц на поверхности металла. Электродвижущая сила Е(э.д.с.)-максимальное напряжение гальванического элемента, отвечающего обратимому протеканию происходящей в нем реакции.
50.Обратимые и необратимые электроды. Уравнение Нернста. Концентрационный гальванический элемент. Анод и катод в концентрационном элементе.
В результате изучения потенциалов различных электродных процессов установлено , что их величины зависят от следующих факторов: 1) от природы веществ –участников электродного процесса. 2) от соотношения м/у концентрациями этих веществ 3) от температуры системы. Эта зависимость выражается уравнением Нернста (В. Нернст, 1889г.): ф=ф0+2,3RT/nF *lg([Ox]/[Red]). Ф0-стандартный электродный потенциал данного процесса – константа, физический смысл которой рассмотрен ниже; R-газовая постоянная; T- абсолютная температура; n- число электронов, принимающих участие в процессе; F- постоянная Фарадея (96500 Кл/моль); [Ox] и [Red]- произведения концентраций в-в, участвующих в процессе в окисленной (Ох) и в восстановленной (Red) формах. Концентрационные элементы состоят из одинаковых электродов, отличающихся активностями потенциалопределяющего иона  Действительно, из уравнения Нернста следует, что при
Действительно, из уравнения Нернста следует, что при  ЭДС концентрационного элемента
ЭДС концентрационного элемента  равна
равна 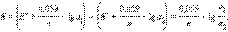 ЭДС этих элементов обычно очень мала. Концентрационные элементы используются при определении pH и концентраций труднорастворимых солей.
ЭДС этих элементов обычно очень мала. Концентрационные элементы используются при определении pH и концентраций труднорастворимых солей.
51.Явление поляризации. Поляризация анодная и катодная. Причины, вызывающие поляризацию электродов (концентрационная и активационная поляризации).
При смещении потенциала электрода в положительную или отрицательную сторону на нём начинает происходить окисление или восстановление. Такое явление смещения называется поляризацией. Иначе говоря поляризация позволяет управлять процессом электролиза и делать электроды анодами или катодами смотря к какому источнику подключать. Поляризация электрода в отрицательную сторону связана с процессом восстановления и наоборот. Процесс восстановления- катодный процесс, процесс окисления – анодный процесс. Чем сильнее поляризован электрод тем с большей скоростью идёт реакция. Величина поляризации необходимая для протекания данного электродного процесса с определённой скоростью называется перенапряжением данного электродного процесса.
52.Коррозия металлов. Классификация коррозионных процессов. Химическая и электрохимическая коррозия.
Коррозия- самопроизвольное разрушение металла, происходящего под хим. воздействием окруж. среды. Хим.коррозия- разрушение металла под действием окислителей окружающей среды, если среда не электропроводна. Электрохимическая коррозия. Возникает в хим. средах с ионной проводимостью при наличии контакта разнородных металлов за счет образования гальванических микроэлементов(Подводные части судов, паровые котлы).
53.Способы защиты металлов от коррозии. Химическая обработка среды.
Химическая обработка среды. Изменение св-в коррозионной среды пригодно для случаев, когда защищаемое изделие эксплуатируется в ограниченном объеме жидкости. Метод состоит в удалении из раствора, в котором эксплуатируется защищаемая деталь, растворенного кислорода(деаэрация) или в добавлении к этому раствору в-в, замедляющих коррозию, - ингибиторов. В зависимости от вида коррозии, природы металла и раствора применяются различные ингибиторы. При атмосферной коррозии применяют хорошо адсорбирующиеся на металле в-ва: моноэтаноламин, карбонат аммония. Уротропин, нитрит натрия. Для нейтральной коррозионной среды и растворов солей в качестве ингибиторов используют неорганические соли хромовых кислот, фосфорной, кремниевой, азотной и азотистой кислот. В кислых средах используют органические ингибиторы, содержащие атомы азота, серы, фосфора, кислорода и группировки атомов с насыщенными связями. Защитные действия ингибиторов обусловлено тем, что их молекулы или ионы адсорбируются на поверхности металла или каталитически снижают скорость коррозии, а некоторые из них(хроматы и дихроматы) переводят металл в пассивное состояние.
54.Способы защиты металлов от коррозии. Защитные покрытия.
Изменение коррозионных св-в металла достигается его легированием или нанесением на поверхность металла защитных покрытий. Из химически стойких сплавов наиболее широкое применение имеют нержавеющие стали, в состав которых входит до 18% хрома и до 10% никеля. Покрытия применяемые для защиты металлов подразделяются на металлические, неметаллические и образованные в результате химической или электрохимической обработки поверхности металла. В качестве металла для покрытия обычно применяют металлы, образующие на своей поверхности защитные плёнки(Cr, Ni, Zn, Cd, Al, Sn, реже Au, Ag). Неметаллические: лаки, краски, эмали. Фенолоформальдегидные и другие смолы. Для длительной защиты от атмосферной коррозии металлических сооружении, деталей, машин, приборов чеще всего применяются лакокрасочные покрытия. Покрытия создаваемые хим. и электрохим. обработкой металла представляют собой защитные оксидные и солевые плёнки. (оксидтрование Al, фосфатирование стальных изделий).
55.Способы защиты металлов от коррозии. Электрохимическая защита. Протекторная защита. Катодная защита.
К электрохимическим методам защиты металлов относятся катодная защита, протекторная защита и др. При катодной защите защищаемая конструкция или деталь присоединяется к отрицательному полюсу источника электрической энергии и становится катодом. В качестве анодов используются куски железа или специально изготовленные сплавы. При надлежащей силе тока в цепи на защищаемом изделии происходит восстановление окислителя, процесс же окисления претерпевает в-во анода. Протекторная защита осуществляется присоединением к защищаемому металлу большого листа, изготовленного из другого, более активного металла – протектора. В качестве протектора при защите стальных изделий обычно применяют цинк или сплавы на основе магния. При хорошем контакте м/у металлами защищаемый металл(Fe) и металл протектора(напр Zn) оказывают друг на друга поляризующее действие. Согласно взаимному положению этих металлов в ряду напряжений, железе поляризуется катодно, а Zn- анодно. В результате этого, на железе идет процесс восстановления того окислителя, который присутствует в воде(обычно растворенный кислород), а Zn окисляется. И протекторы и катодная защита применимы в средах, хорошо проводящих электрический ток, например в морской воде. В частности, протекторы широко применяются для защиты подводных частей мрских судов.
57.Химическое взаимодействие металлов с растворами щелочей. Приведите примеры соответствующих уравнений реакций.
Взаимодействие Ме с растворами щелочей. Щелочами металлы окисляться не могут, так как щелочные металлы являются одними из наиболее сильных восстановителей. Поэтому их ионы одни из наиболее слабых окислителей и в водных р-рах практических свойств окислителя не проявляют. Однако в присутствии щелочей окисляющее действие воды может проявиться в большей мере, чем в их отсутствии. При окислении металлов водой образуются гидроксиды и водород. Если оксид и гидроксид относится к амфотерным соединениям, то они будут растворяться в щелочном р-ре. В результате пассивные в чистой воде металлы могут энергично взаимодействовать с растворами щелочей:
| H2O (окислитель H+)+щелочь(например, NaOH ) | ||
| Активные | Средней активности | Малоактивные |
| Реагируют только Be | Реагируют Al, Zn, Sn, Pb | Не реагируют |
Zn+2HOH+2NaOH=Na2[Zn(OH)4]+H2|-
58.Химическое взаимодействие металлов с водой и с растворами солей. Объясните закономерности и приведите примеры соответствующих уравнений реакций.
| H2O (окислитель H+) | ||
| Активные | Средней активности | Малоактивные |
| Реагируют | Реагируют, пассивируются: Al, Ti, Cr, Fe, Co, Ni, Zn, Sn, Cd, Pb | Не реагируют |
Ca+2H2O=Ca(OH)2+H2|-
Me+соль:
1. металлы более активные вытесняют менее активные из их солей Zn+CuSO4=ZnSO4+Cu
2. Взаимодействие металла с солями, дающими кислую реакцию среды засчет гидролиза металл., происходит также, как в растворе соответствующей к-ты, если металл, образующий соль, активнее растворяющего металла и кислорода.
Если соль не подвергается гидролизу или дает щелочную реакцию среды, то металл окисляется кислородом, растворенным в электролите. Fe в р-ре AlCl3: AlCl3+2H2O=AlOHCl2+HCl 2Fe+2HCl=FeCl2+H2|-