Вторичные коллагенозы.
Гомоцистинурия – наследственное заболевание, связанное с нарушением обмена серосодержащих аминокислот
Тип наследования: аутосомно-рецессивный. Патологический ген находится в 21 хромосоме (21q22).
Причина: дефицит фермента цистатионсинтетазы, в результате нарушается образование цистатионина из серина и гомоцистина в головном мозге. Таким образом, концентрация цистатионина резко снижается, а содержание гомоцистина в крови, моче и ликворе возрастает. Гомоцистин способен активировать факторы свёртывания (развивается тромбофилия), изменять структуру коллагена (характерная клиника). В 20-40% случае введение больших доз витамина В6, являющегося кофактором фермента цистатионсинтетазы, приводит к восстановлению его активности и нормализации экскреции гомоцистина (пиридоксинзависимая форма).
Патоморфология: утолщение интимы и фиброз сосудистой стенки, множественные разной давности инфаркты головного мозга, тромбоз синусов твёрдой мозговой оболочки.
Клиника сходна с синдромом Марфана:
o Более тяжелые поражения ЦНС с 5-9 месяцев (снижение интеллекта, судорожный синдром)
o Поражение сосудов (варикоз, тромбофлебит, артериальные и венозные тромбозы, тромбоэмболии)
o Поражение глаз: подвывих хрусталика к 3-10 годам, вторичная глаукома, катаракта
o Костно-скелетные нарушения (ранний остеопороз, вальгусные голени, сколиоз, «куриная» грудная клетка, плоскостопие, «полая» стопа, разболтанность суставов, арахнодактилия через несколько лет после рождения)
o Поражение кожи и её производных: светлые, редкие и ломкие волосы, множественные эритематозные пятна на щёках
Диагностика: а) для определения в моче гомоцистина проводится цианиднитропруссидная реакция; б) увеличение в крови количества гомоцистина и метионина; в) определение специфического ферментативного дефекта в фибробластах.
Лечение: 1) диета, обогащенная цистином с ограничением метионина, 2) большие дозы витаминов группы В (В6 500-1200 мг/сут) при пиридоксинзависимой форме, бетаин (триметилглицин) при пиридоксинустойчивой форме, 3) фолиевая кислота, 4) препараты кальция, аскорбиновой кислоты.
УМСТВЕННАЯ ОТСТАЛОСТЬ
Умственная отсталость - это стойкое нарушение психического развития, обусловленное органическим поражением головного мозга или его недоразвитием.
В 70 % случаев она генетически обусловлена.
Критерии умственной отсталости.
1. Снижение IQ менее 70.
2. Недостаток социальной компетенции индивида.
3. Недостаточность познавательной деятельности.
4. Нарушение абстрактного мышления.
5. Инертность психических процессов.
По этиопатогенетическим признакам умственная отсталость классифицируется:
1.Эндогенно-обусловленная:
• хромосомные нарушения;
• генные нарушения;
• мультифакториальные;
2.Экзогенно-обусловленная (различные факторы внешней среды, воздействующие на плод).
Различают около 1000 наследственных заболеваний, протекающих с умственной отсталостью. Из них примерно:
- 300 синдромов обусловлены мутацией аутосом;
- 50 синдромов - мутацией половых хромосом;
- 600 синдромов являются болезнями обмена веществ.
Учитывая вышеизложенное, следует подчеркнуть, что умственная отсталость является генетически гетерогенным состоянием, обусловленным большим числом генных, хромосомных и мультифакто-риальных нарушений и широкого спектра внешнесредового воздействия
Разработаны методы ранней диагностики нарушений обмена веществ, протекающих с умственной отсталостью (скрининг-программы) и методы селективного цитогенетического скрининга новорожденных.
Клиника нарушений обмена веществ, протекающих с умственной отсталостью изложена в соответствующих главах, ниже рассмотрим синдромы, не вошедшие в другие главы.
1) Синдром Корнелии де Ланге.
Тип наследования неизвестен, возможна полигенная наследственность. Большинство случаев в родословной имеет спорадический характер. Отмечается деления 3 или 8 хромосомы. Частота 1:30 000-50 000 населения.
Клиника:
· Брахимикроцефалия
· Типичное лицо: синофриз (сросшиеся брови), длинные ресницы, вывернутые наружу ноздри, запавшая переносица, длинный фильтр (увеличенное расстояние между основанием носа и краем верхней губы)
· Гипертрихоз, гирсутизм, низкий рост волос на лбу и затылке
· Голубоватый оттенок кожи вокруг глаз, носа, губ вследствие усиленного венозного рисунка
· Малые кисти и стопы, клинодактилия (медиальное или латеральное искривление пальцев кисти, чаще 4й,5й, стопы, чаще 1й) или камптодактилия (сгибательная контрактура пальцев кисти, чаще 5ого). Контрактуры локтевых суставов
· Отставание в росте
· Умственная отсталость различной степени выраженности
Диагностика: а) клинические данные, б) на рентгенограммах: конусовидные эпифизы, гипоплазия головки лучевой кости, горизонтальное расположение рёбер.
2) Синдром Коффина-Сириса.
Тип наследования неизвестен. В основном, возникает спорадически.
Клиника:
· пренатальная гипотрофия с отставанием в росте
· умеренная или тяжёлая диффузная мышечная гипотония
· умеренная микроцефалия, гидроцефалия
· грубые черты лица (густые брови, полные губы, гипотелоризм, птоз, преаурикулярные выросты)
· скелетные аномалии: гипо - или аплазия (отсутствие) 5го пальца и ногтевой пластины на стопах и кистях (ногти на пальцах кистей гипоплазированы в меньшей степени), аномалии позвонков, расщелина нёба, разболтанность суставов с подвывихом лучевой кости в локтевом суставе
· редкие волосы на голове в сочетании с общим гирсутизмом
· крипторхизм
· врождённые пороки сердца
· глубокая умственная отсталость
Диагностика: а) клиничексие симптомы; б) на рентенограммах: coxa valga, гипоплазия ключиц, надколенника, отставание костного возраста
3) Синдром Коффина-Лоури.
Описан в 1966 году. Тип наследования: Х-сцепленный доминантный. Клиника в большей степени выражена у мужчин.
Клинически:
- Типичное лицо: антимонголоидный разрез глаз, выступающие надбровные дуги, гипертелоризм, широкий нос с толстыми крыльями
- Костно-скелетные аномалии: низкий рост, кифосколиоз, килевидная грудная клетка, конусовидные пальцы, кисти широкие, большие; мягкие, гиперподвижные суставы
- Поперечная борозда на ладони
- Умственная отсталость различной степени выраженности.
4) Синдром Рубинштейна-Тейби.
Описан в 1963 году. Тип наследования неизвестен. Частота 1 на 25000 населения.
Клиника:
Ø Снижение роста, отставание костного возраста
Ø Черепно-лицевыми аномалиями: микроцефалия, выступающий лоб с низким ростом волос, полуптоз, страбизм (косоглазие), длинныме ресницы, антимонголоидный разрез глаз, гипертелоризм, гримаса, напоминающей улыбку, высокое нёбо, клювовидный нос, микрогнатия, аномалии прикуса изубов, низко расположенные и деформированные ушные раковины
Ø Костно-скелетные аномалии: широкие терминальные фаланги I пальцев кистей и стоп, брахидактилия (короткие пальцы), полидактилия (увеличенное число пальцев), гиперподвижность суставов, дефекты развития позвоночника, грудины, рёбер
Ø Крипторхизм
Ø Нарушение зрения (катаракта, гиперметропия, колобома радужки, атрофия зрительных нервов)
Ø Различные пороки внутренних органов
Ø Умственная отсталость различной степени выраженности.
5)Синдром Мартина-Белл (синдром фрагильной (ломкой) Х-хромосомы).
Тип наследования: Х-сцепленный рецессивный. Частота 1:1 000- 1:2000 новорождённых мальчиков. Основная причина: экспансия тринуклеотидов ЦГГ (цитозин-гуанин-гуанин) в длинном плече Х-хромосомы (вторичная перетяжка), что приводит к недостаточной экспресиии белка FMR 1, нобходимого для нормального развития нервной системы. В норме число повторов ЦГГ 29-31. При количестве повторов 55-200 заболевание клинически не проявляется, при количестве повторов превышающих 200 возникает полная клиническая картина.
Клиника:
ü Большая масса при рождении 3,5- 4 кг
ü Макроорхизм без эндокринной патологии
ü Внешний вид: макроцефалия, высокий и широкий лоб, длинное лицо, увеличенный подбородок, тупой клювовидно загнутый кончик носа, большие оттопыренные низко расположенные уши, широкие кисти и стопы
ü Патология соединительной ткани: гипермобильность суставов и гиперэластичная кожа
ü Умственная отсталость различной степени выраженности
ü Светлая радужка, светлые волосы
ü Неврологические симптомы: умственная отсталость, мышечная гипотония, дискоординация в движениях, глазодвигательные, пирамидные и экстрапирамидные движения. Быстрая сбивчивая речь с эхолалией и персеверациями.
ü Психические нарушения: агрессивность, двиагтельная расторможенность, шизофреноподобная симптоматика (подрыгивают, похлопывают руками, поворачиваются вокруг собственной оси, осуществляют «манежный» бег, гримасничают), аутизм
Диагностика: а) клинические данные; б) эндонуклеазная рестрикция и саузерн блоттинг (длинное плечо Х-хромосомы выглядет под микроскопом суженной, перетянутой, хрупкой – отсюда название синдрома).
6)Синдром Ангельмана ("счастливой куклы").
Тип наследования: аутосомно-рецессивный, большинство случаев ноят спорадический характер. Доказан геномный импритинг: наблюдается микроделеция 15 хромосомы (15q11-q13) материнского происхождения или однородительская отцовская дисомия. Описан синдром в 1965 году.
Клиника:
§ Задержка психомоторного развития, ведущая к тяжелой умственной отсталости с грубой задержкой речевого развития
§ Характерный внешний вид: микробрахицефалия, прогени, макростомия, широкие межзубные промежутки
§ Неврологические симптомы: судороги, атаксия с необычной походкой (на широко расставленных ногах с согнутыми в локтях руками – походка «механической куклы»), мышечная гипотония, повышение сухожильных рефлексов, приступы немотивированного спонтанного смеха, расходящееся косоглазие, хореоатетоидные гиперкинезы с постоянным высовыванием языка и стереотипными движениями рук
§ В некоторых случая отмечается гипопигментация кожи и волос
7) Синдром Лоуренса – Муна – Барде - Бидля.
Описан впервые в 1864 году офтальмологом Гэрингом, а в последующем Стором в 1865 году, Лоуренсом и Муном в 1866 году, которые диагностировали у 4-х сибсов ожирение, умственную отсталость, пигментный ретинит, спастическую параплегию. В 1920 году Барде наблюдал похожих больных, у которых помимо перечисленных симптомов нередко обнаруживали также полидактилию. В 1925 году отдельные наблюдения объедиилм в одну нозологическую единицу, имеющую генетически гетерогенные варианты. Тип наследования: аутосомно-рецессивный. Выделяют три клинических варианта: полный, частичный, стёртый.
Клиника:
o ожирение, которое появляется на 1-м году жизни и прогрессирует
o гипогонадизмом (гипогенитализм) или задержка полового созревания
o пигментная дегенерация сетчатки, носящей прогрессирующий характер и ведущей к полной потере зрения
o врожденная патология почек и сердца
o врождённые пороки развития головного мозга (атрофия извилин, гидроцефалия, асимметрия полушарий, агенезия мозолистого тела) и умственная отсталость различной степени выраженности
o неврологические симптомы: спастическая параплегия, судороги, мозжечковая атаксия, экстрапирамидные нарушения
o костные аномалии: постаксиальная полидактилия
Диагностика: а) клинические симптомы; б) ЭЭГ: грубые нарушения биоэлектрического возрастного ритма; в) ЭхоЭГ: расширение III и боковых желудочков; г) УЗИ сердца, органов брюшной полости, почек: врождённые аномалии развития почек и сердца.
8) Синдром Маршалла-Смита.
Тип наследования: точно неизвестен, возможно, аутосомно-рециссивный. Чаще встречаются спорадические случаи. Описан в 1971 году.
Клиника:
Ø С рождения отмечается ускоренный рост и созревание скелета
Ø Типичное лицо: плоские орбиты, высокий выступающий лоб, вздёрнутый нос, экзофтальм, голубые склеры, микрогнатия (недоразвитая нижняя челюсть), гипоплазия средней части лица
Ø Костно-скелетные аномалии: макроцефалия, широкие проксимальные и средние фаланги пальцев рук и узкие дистальные фаланги
Ø Задержка психомоторного развития в виде умственной отсталости различной степени выраженности
Ø Гипертрихоз
Ø Пупочные грыжи
Ø Диффузная мышечная гипотония, врождённые пороки развития головного мозга (макрогирия – широкие извилины, атрофия мозгового вещества, агенезия мозолистого тела)
Ø Атрезия или стеноз гортани, в результате может нарушаться глотание, и дети отстают в весе
Ø Прогноз зависит от тяжести поражения дыхательной системы (стридор, пневмония, ателектазы).
Приложение
Генеалогический метод диагностики генетических заболеваний:

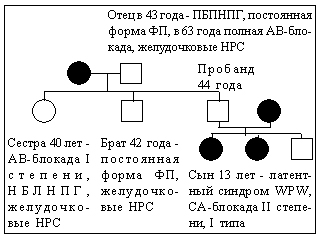
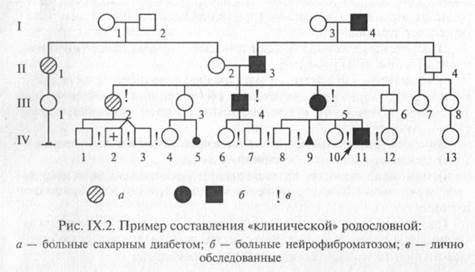

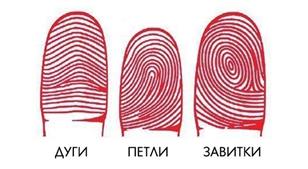 Дерматоглифический метод диагностики генетических заболеваний
Дерматоглифический метод диагностики генетических заболеваний

Синдром Шерешевского-Тернера XO
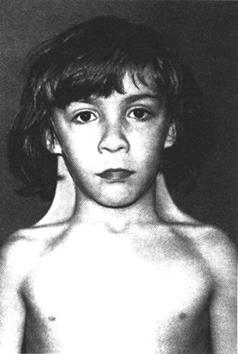

Синдром Дауна:


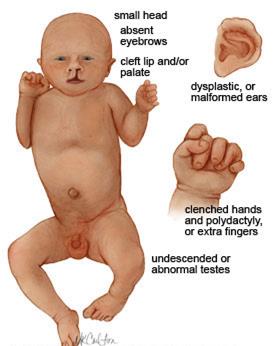
 Синдром Патау
Синдром Патау
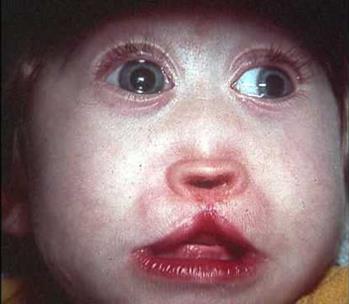
 синдром эдвардса
синдром эдвардса


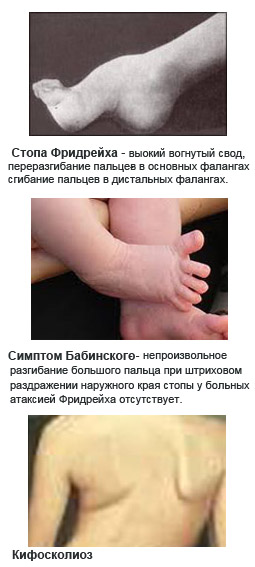 атаксия фридрейха
атаксия фридрейха
Болезнь Рефсума:
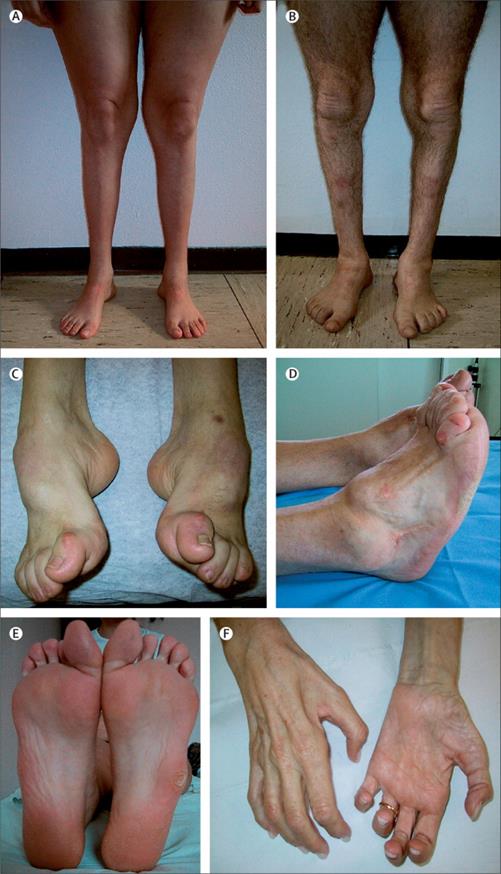
Болезнь Рефсума (см.выше и далее) 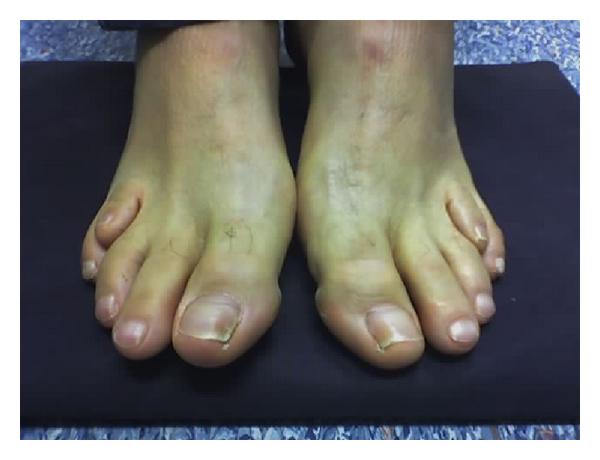

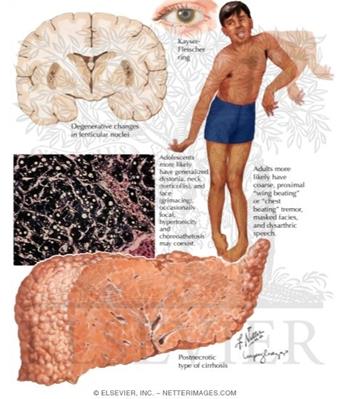
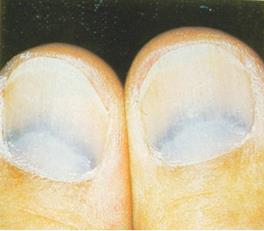 Болезнь Вильсона-коновалова:
Болезнь Вильсона-коновалова:

Хорея Гетингтона:

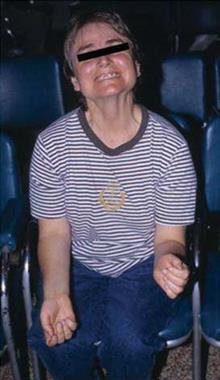
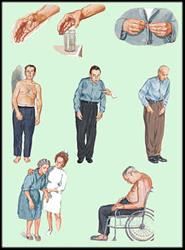

Б-нь Паркинсона:
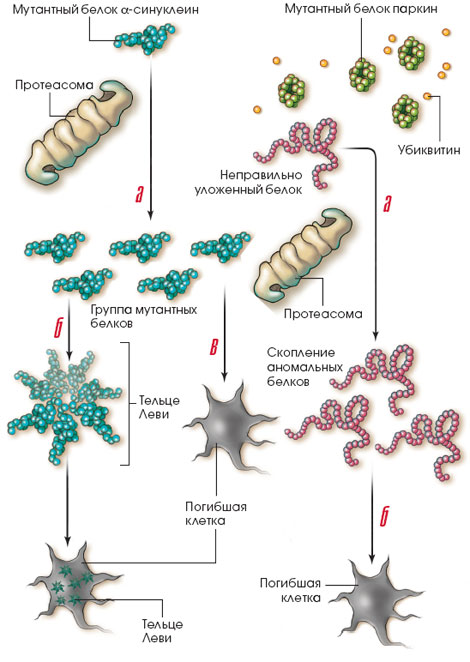
БАС:

Болезнь Дюшена
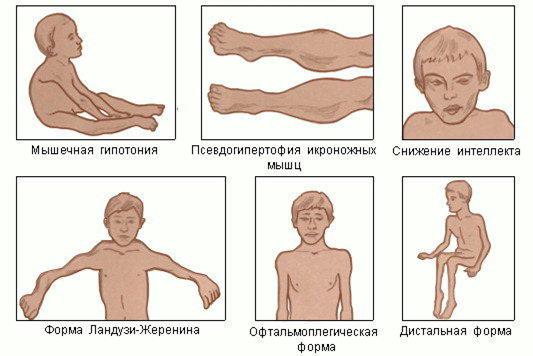

болезнь Эрба-Рота:
Немалиновая миопатия:
Миотония Томсена:
Спинальная амиотрофия Верднига-Гоффмана:

Амиотрофия Шарко-Мари-Тута:
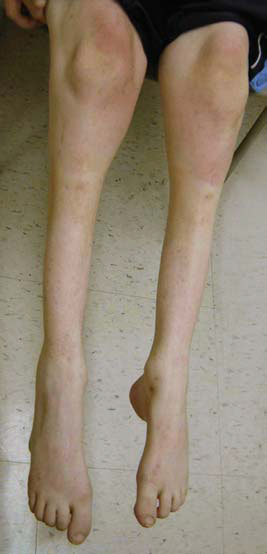
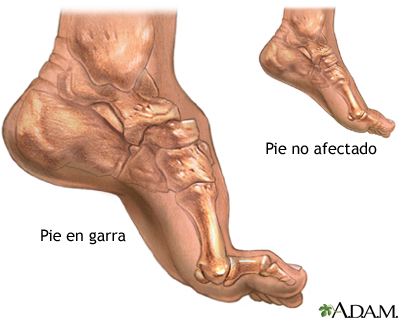
Миастения гравис:

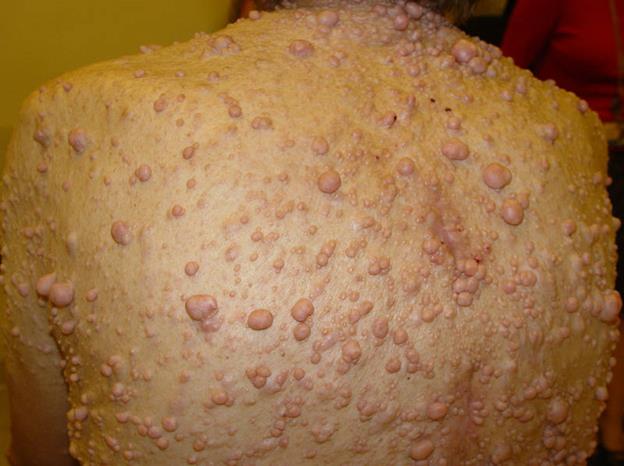
Нейрофиброматоз Реклингаузена:
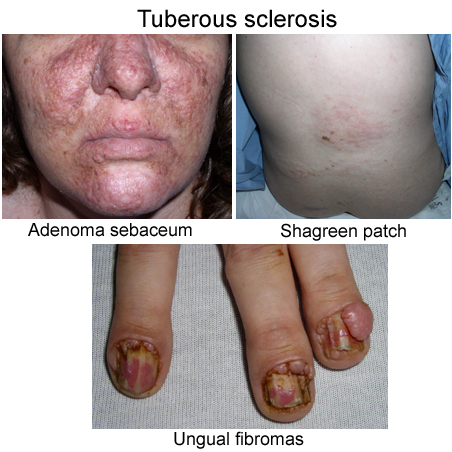
Туберозный склероз:
Синдром недержания пигмента Блоха-Сульцбергера:
Энцефалотригеминальный ангиоматоз Штурге (Стурге, Стурдже) –Вебера:



 Атаксия – телеангиэктазия Луи-Бар:
Атаксия – телеангиэктазия Луи-Бар:
 Галактоземия:
Галактоземия:
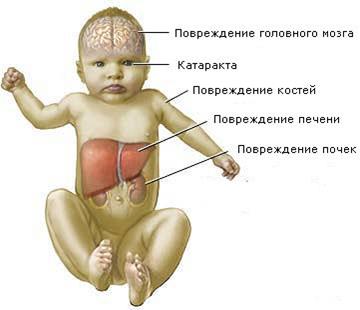
Гликогеноз Гирке и Помпе:

Глобоидно-клеточная лейкодистрофия Краббе-Бенеке:

Адренолейкодистрофия:


Болезнь Тея-Сакса:


Болезнь Фабри:

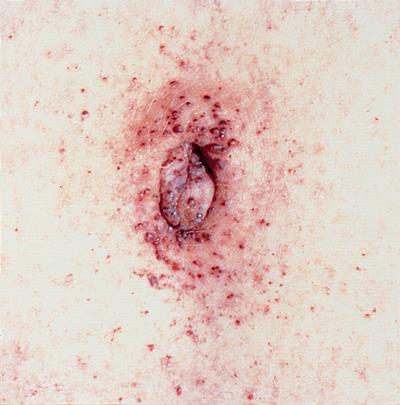
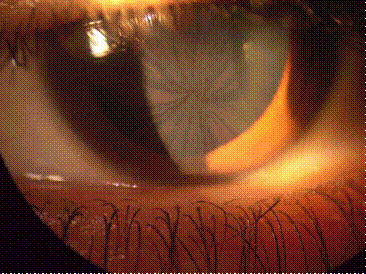
Болезнь Ниманна-Пика:

Фенилкетонурия:
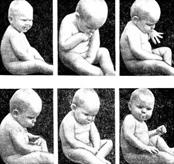
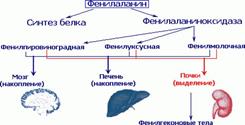
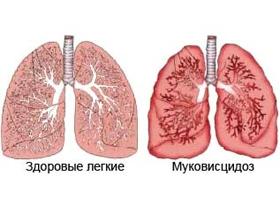
Муковисцедоз:
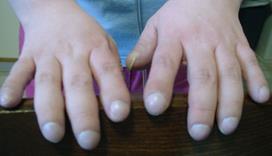

Целиакия:
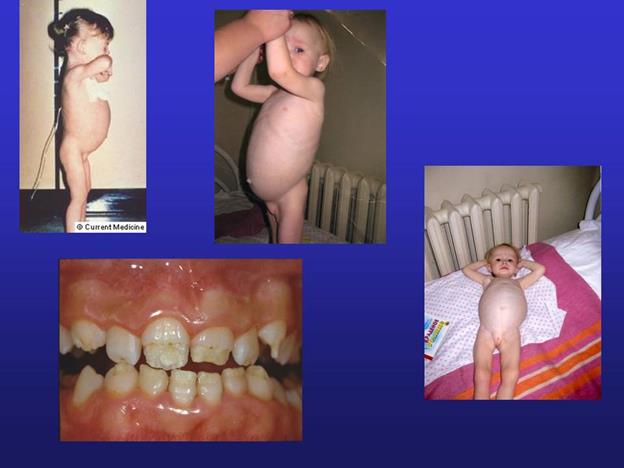

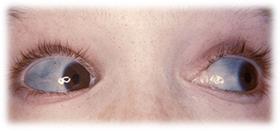
Болезнь Марфана:

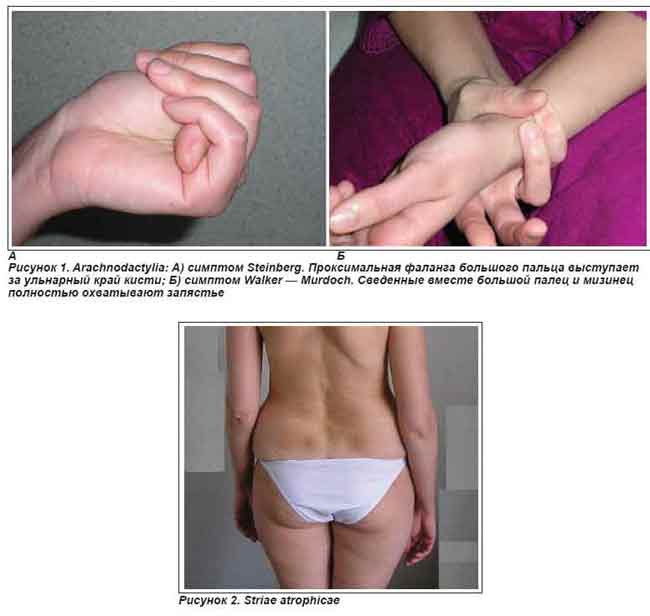

Болезнь Элерса-Данлоса:

Несовершенный остеогенез:


Синдром Лоуренса-Муна-Барде-Бидля:
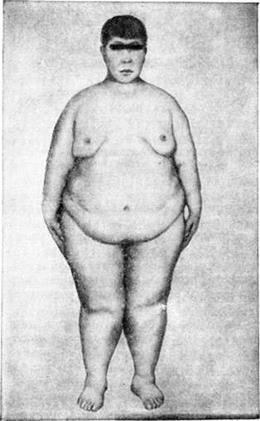
Умственная отсталость:


