А Основные методы современной биотехнологии. Методы секвенирования ДНК, его основные принципы, цели.
К основным методам биотехнологии относятся:
1. ПЦР (полимеразная цепная реакция)
2. Анализ длины рестрикционных фрагментов
3. Сверхэкспрессия белков
Секвенирование - последний этап молекулярного анализа предварительно отобранного, клонированного и протестированного более простыми методами фрагмента ДНК.
Секвенирование представляет собой определение нуклеотидной последовательности фрагмента ДНК путем получения серии комплементарных молекул ДНК, различающихся по длине на одно основание.
Существует два основных метода секвенирования: метод Максама-Гилберта (основан на химическом расщеплении ДНК по одному основанию) и метод Сэнгера (дидезокси-метод). Метод Сэнгера более надежен и прост в исполнении, и на практике его используют чаше.
Фредерик Сэнгер (1918-2013). Две Нобелевские премии: за расшифровку структуры инсулина (1958), и за открытие метода секвенирования ДНК (1980)& Метод Сэнгера или дидезоксисеквенирование основан на синтезе изучаемой цепи ДНК in vitro с остановкой синтеза на заданном основании путем присоединения дидезоксинуклеотида. Дидезоксинуклеотид лишен гидроксильных групп при атомах сахарного кольца не только в 2'-, но и в З'-положении, что делает его неспособным формировать фосфодизфирную связь со следующим нуклеотидом. Такие дидезоксинуклеотиды получают путем синтеза.
Стадии секвенирования ДНК классическим «методом терминаторов» по Ф. Сэнгеру:
1. Клонирование, а затем гибридизация изучаемого фрагмента ДНК с праймером,
2. Ферментативный синтез ДНК в 4 пробирках с ДНК-полимеразой, 4 нуклеотидами, и радиомеченым ddNTP-терминатором
3. Денатурацию полученных продуктов
4. Электрофорез в полиакриламидном геле на 4-х дорожках (по числу типов нуклеотидов)
5. Анализ результатов на радиоавтографе. Обычно можно различить 250—350 полос, т.е. прочитать последовательность в 250-350 п.н.
Таким образом, по размеру синтезированных фрагментов может быть определена локализация дидезоксинуклеотидов и порядок соответствующих им нуклеотидов в исходной молекуле ДНК.
Для определения нуклеотидной последовательности (т. е. первичной структуры) конкретного района ДНК в первую очередь необходимо упростить ее, что достигается путем разрезания ее на относительно короткие фрагменты. Сделать это можно, например, с помощью специальных «скальпелей» для ДНК — ферментов рестриктаз, о которых уже шла речь выше.
При секвенировании простейших организмов, у которых геном относительно невелик, обычно используют процедуру, называемую условно «сверху вниз». Всю ДНК «разрезают» на кусочки с помощью уже упоминавшихся выше ферментов рестриктаз, затем секвенируют эти кусочки по отдельности, а после «склеивают» из них полный геном. «Склеивание» оригинала осуществляется за счет того, что нуклеотидные последовательности разных кусочков перекрываются друг с другом, т. е. одинаковы по концам. Эта методология получила название «дробовика».
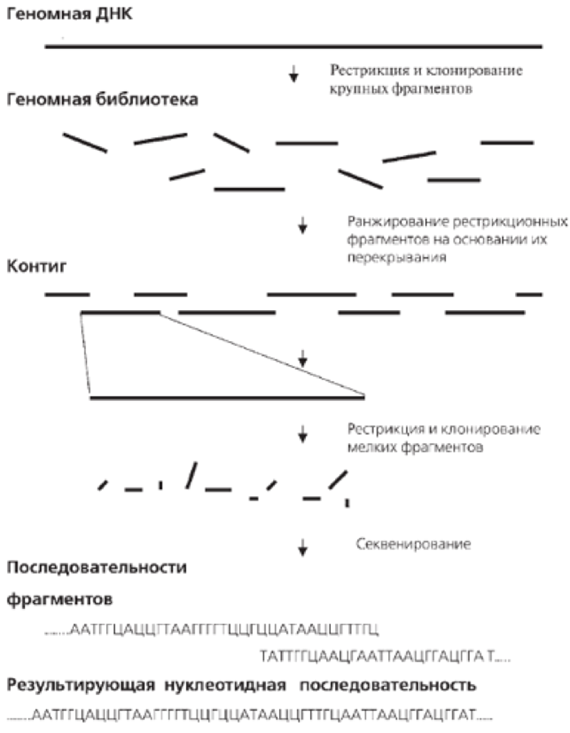
Для прочитывания более длинных последовательностей существует ряд методов, представляющих собой разные модификации метода Сэнгера. Эти методы основаны на предварительном клонировании ДНК в векторах, сконструированных на основе фага М13 E.coli для получения протяженных одноцепочечных участков ДНК, которые могут быть непосредственно секвенированы без денатурации и праймирования. Особенность фага М13 E.coli состоит в возможности его существования в двух формах: двухцепочечной репликативной, функционирующей как плазмида, и одноцепочечной фаговой, использующейся в качестве матрицы для секвенирования. Выделив после клонирования одноцепочечные фаговые ДНК со вставкой (размер около 500 п.н.), праймер гибридизуютс последовательностью вблизи вставки, и проводят дидезоксисеквенирование.
Для секвенирования крупных фрагментов ДНК (около 2000 п,н.) используют более сложные (комбинированные) подходы. Один из них заключается в предварительном клонировании данного фрагмента в плазмилном векторе и построении его подробной рестрикционной карты, идентификации перекрывающихся рестрикционных фрагментов длиной 100-500 п.н. Далее, субклонировав каждый из этих фрагментов в ДНК M13 E.coli, их секвенируют и определяют последовательность исходного участка ДНК. Так как субклонированные фрагменты могут быть встроены в противоположных направлениях, то праймер в одном случае будет инициировать синтез первой цепи, а вдругом - комплементарной ей, а значит возможно одновременное секвенирование обеих цепей.
Для секвенирования фрагментов ДНК более 5000 п.н. используют методы секвенирования двухцепочечных плазмидных ДНК, не требующие субклонирования. Один из них носит название «праймер-опосредованной прогулки» («блуждающей затравки»). Суть метода заключается в последовательном дидезоксисеквенировании по 250-350 п.н. Этапы метода представляют собой цепочку циклов, включающих синтез праймера, дидезоксисеквенирование и идентификацию нуклеотидной последовательности: 1) отжиг плазмидной ДНК, содержащей вставку, спраймером, комплементарным последовательности одной из цепей векторной ДНК; дидезоксисеквенирование и идентификация первых 250-350 п.н. вставки; 2) синтез второго праймера, комплементарного сегменту вставки, который отстоит от места связывания первого праймера примерно на 300 п.н., и секвенирование следующих 250-350 п.н., и так далее, пока не секвенируют весь фрагмент.
Обязательное условие при секвенировании очень длинных фрагментов иметь праймер длиной не менее 24 нуклеотидов. Это необходимо для того, чтобы избежать спаривания праймера внутри вставки более одного раза. Этим методом были секвенированы фрагменты ДНК, клонированные в бактериофаге А. (=20 т.п.н.) и космидном векторе (=40 т.п.н.).
В настоящее время широкое распространение получила методология автоматического ДНК-секвенирования с использованием меченных различными флуорохромамидиде-эоксинуклеотидов - электрофорез в этом случае проводят на одной дорожке и на элект-рофореграмме каждому из нуклеотидов соответствует свой цвет полосы, которую сканируют в луче лазера, занося данные в компьютер, который сопоставляет их и идентифицирует, выводя на экран нуклеотидную последовательность.. Этот метод широко применялся в ходе реализации программы «Геном человека». Использование автоматических секвенаторов снизило стоимость секвенирования одного звена с 1 $ до 0,1 $. Однако существуют еще более быстрые методы автоматического секвенирования. Один из них - секвенирование путем гибридизации исследуемой последовательности ДНК с набором олигонуклеотидов (олигонуклеотидной матрицей), включающим все возможные варианты перестановок из 4 стандартных нуклеотидов (А, G, С, Т) определенной длины. Наиболее удобными считаются наборы матриц (чипы) из октануклеотидов., при этом количество возможных вариантов нуклеотидов составляет 65536. Секвенируемый фрагмент ДНК, меченный радиоактивным фосфором, гибридизуется только с комплементарными его участкам октануклеотидами. В результате определяется спектр октануклеотидов, составляющих исследуемый фрагмент ДНК. Локализация октамеров в изучаемом фрагменте ДНК устанавливается при помощи специальной компьютерной программы.