V гетерогенных хим. реакций.
Многие химические процессы, имеющие большое значение в технике, относятся к числу гетерогенных реакций: горение твердого и жидкого топлива, хим. и электрохим. коррозия металлов и сплавов. Реакция в гетерогенной системе осуществляется на поверхности раздела фаз. В связи с тем что поверхность раздела фаз является реакционным пространством гетерогенной хим. реакции, концентрацию газообразных и жидких веществ, участвующих во взаимодействии, измеряют кол-вом молей этих веществ, приход. на единицу реакционной поверхности (моль/м2 или моль/см2), и наз. поверхностной концентрацией CS.
Скорость: средняя(V=±DСS/Dt) и истинная (или мгновенную) скорость (VИСТ=±dCS/dt) реакции - измеряется изменением поверхностной концентрации одного из веществ (газа или жидкости), участвующих в реакции, за единицу времени; размерность скорости гетерогенной реакции моль/(м2*с) или моль/(см2*с).
Зависимость скорости гетерогенной химической реакции от поверхностной концентрации реагентов, так же как и зависимость скорости гомогенной реакции от объемной концентрации, определяется законом действующих масс  .
.
Скорость гетероген. взаим. часто снижается из-за того, что молекулы газообразного (или растворенного) реагента не успевают поступать из объема к реакционной поверхности. Чтобы скорость реакции при заданной температуре была максимальной, необходимо обеспечить интенсивный подвод реагента из объема V к реакционной поверхности S. Стадия подвода реагента к поверхности раздела фаз часто является лимитирующей стадией гетерогенной реакции.
Скорость взаим., протекающего в гетер. системе, зависит и от состояния реакционной поверхности, которое во многом опр. интенсивностью отвода от поверхности продуктов реакции. Последние иногда резко искажают свойства реакционной поверхности, изменяя ее природу.
Следует отметить, что при T = const на единице поверхности раздела фаз число столкновений молекул постоянно.
№21. Зависимость V реакции от концентрации. Закон действующих масс.
Закон действующих масс.В 1865 г. проф. H. H. Бекетов впервые высказал гипотезу о количественной взаимосвязи между массами реагентов и временем течения реакции: «...притяжение пропорционально произведению действующих масс». Эта гипотеза нашла подтверждение в законе действующих масс, который был установлен в 1867 двумя норвежскими химиками К.Гульдбергом и П. Вааге. Современная формулировка закона действующих масс такова: скорость гомогенной хим. реакции при постоянной температуре реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции.
Для реакции: аА + bВ ® mM + nN
математическое выражение закона действующих масс имеет вид:
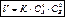
где V — скорость реакции; K — коэффициент пропорциональности, наз. константой скорости хим. реакции (при Ca= Cb= 1 моль/л Kчисленно равна V); Ca и Cb — концентрации реагентов А и В; а, Ь — стехиометрические коэфф. в уравнении реакции.
Константа V химической реакции k определяется природой реагирующих веществ и зависит от температуры, от присутствия катализатора, но не зависит от концентрации веществ, участвующих в реакции.
Закон действующих масс справедлив только для наиболее простых по своему механизму взаимодействий, протекающих в газах или в разбавленных растворах.
Часто уравнение реакции не отражает ее механизма. Например, запись
аА + bB ® AaBb может быть суммарным ур. сложного взаимодействия, протекающего по стадиям:
A + B®AB
A + AB ®AаB
АаВ+(b-1)B®АаBb
Сложные реакции могут быть совокупностью параллельно или последовательно протекающих процессов. Закон действующих масс справедлив для каждой отдельной стадии реакции, но не для всего взаимодействия в целом. Та стадия процесса, скорость которой минимальна, лимитирует скорость реакции в общем. Поэтому математическое выражение закона действующих масс, записанное для самой медленной (лимитирующей) стадии процесса, приложимо одновременно и ко всей реакции в целом. Если в реакции участвуют два или более веществ, то скорость реакции иногда зависит от концентрации только одного из них и не зависит от концентрации других.
№22. Зависимость V хим. реакции от t0. Правило Вант-Гоффа, Арениуса. Энергия и энтропия активации.
Правило Вант-Гоффа. Скорость химической реакции зависит от температуры, причем при повышении температуры скорость реакции увеличивается. Голландский ученый Якоб Вант-Гофф показал, что при повышении температуры на десять градусов скорость большинства реакций увеличивается в 2—4 раза  где VT1и VT2скорости реакции при температурах T2 и T1; g=VT2/VT1— температурный коэффициент скорости реакции, показывает, во сколько раз увеличивается скорость реакции при повышении температуры на 10 K.
где VT1и VT2скорости реакции при температурах T2 и T1; g=VT2/VT1— температурный коэффициент скорости реакции, показывает, во сколько раз увеличивается скорость реакции при повышении температуры на 10 K.
Уравнение Аррениуса. Функ. зависимость константы скорости хим. реакции k от температуры установил шведский ученый С. Аррениус (1889):

где А-предэкспоненциальный множитель; Еа-энергия активации реакции; T-абс. температура, R-универсальная газовая постоянная=8,31,DSa-энтропия активации.
В уравнение Аррениуса входят две величины: Еа и А. Их физический смысл вытекает из следующих рассуждений. Необходимым условием начала хим. взаим. между двумя молекулами должно быть их соударение. Однако не все соударения молекул заканчиваются актом хим. взаим., т. е. не все соударения эффективны. Более того, доля эффективных соударений от их
общего числа, как правило, незначительна. В этом легко убедиться, вычислив на основании кинет. теории газов возможное число соударений молекул и соответствующую этому числу скорость реакции, а затем сравнить ожидаемую (в расчете на 100%-ную эффективность) скорость с действительной; первая будет во много раз больше последней. Аррениус высказал гипотезу о том, что хим. взаимодействие осущ. только между теми соудар. молекулами, кот. достигли опр. энерг. уровня, характерного для данной реакции, ее энергетического барьера. Если считать такие молекулы активными, то эффективные столкновения происходят только между активными молекулами.
Энергия активации Еа-один из основных параметров, который характеризует скорость хим. взаимодействия, равна той min энергии кот. необходимо сообщить частицам реаг. веществ чтобы они при столкновении могли вступить в хим. взаимодействие, кот. возможно лишь в случаи если будут преодолены силы отталкивания эл. оболочек. Еа<40кДж/моль – реакция быстрая, Еа>120-медленная. Энергия активации необходима в основном для ослабления хим. связей в исходных веществах и для преодоления отталкивания между эл., кот. возникает при сближении молекул и атомов взаим. веществ и мешает их столкновению.
Энтропия активации DSa- учитывает вероятность благоприятной ориентации соударяющихся молекул, т.е. вероятность их столкновения активными участками.
№23. Влияние катализаторов на V реакции. Гомо- и гетерогененный катализ.
Катализаторы — вещ-ва, резко увелич. скорость реакции, но не расходующихся в результате ее протекания. Если катализатор и взаим вещества образуют однофазную систему, катализ наз гомогенным. Если же катализатор нах в системе в виде самостоятельной фазы, катализ называют гетерогенным.
Катализ — наиболее эффективный метод интенсификации промышленных хим процессов. Ферменты — исключительно акт катализаторы; их действие отличается высокой избирательностью. Теория каталитических реакций исходит из некоторых общих положений:
а) катализ как метод изменения скорости реакции применим только тогда, когда энергия Гиббса взаим при данных условиях отрицательна (DG<0);
б) в присутствии катализатора изменяется механизм хим реакции, она протекает через новые стадии, каждая из которых характеризуется невысокой энергией активации.
в) при катализе не изменяется тепловой эффект реакции;
г) если катализируемая реакция обратима, катализатор не влияет на равновесие, не меняет константы равновесия и равновесных концентраций компонентов системы. Он в равной степени ускоряет и прямую, и обратную реакции, тем самым сокращая время достижения равновесия;
д) катализаторы обычно действуют избирательно, селективно.
Активность того или иного катализатора может быть резко повышена или понижена различными добавками. Добавки, увелич. активность катализатора, называются промоторами. Активность, селективность и срок службы катализатора во многом зависят от температуры проведения каталитической реакции.
Гомогенный катализ. (Сабатье и H. Д. Зелинского). Хим взаим катализатора с исходными вещ-вами направ реакцию по пути, отличному от того, что осущ в отсутствие катализатора. В результате этого уменьшается энергия активации протекающего взаимодействия и увеличивается его скорость. В общем виде схему гомогенной каталитической реакции можно представить себе так:
в отсутствие катализатора AàB
в присутствии катализатора K
A+ Kà [AK] (первая стадия),
[AK]àВ +К (вторая стадия).
(Пример: разложения пероксида водорода в присутствии растворенного иода).
Гетерогенный катализ - начинается с адсорбции молекул исх веществ на поверхности твердого катализатора. При этом только обратимая адсорбция является началом каталитической реакции. Необратимая хемо-сорбция приводит к образованию на поверхности катализатора устойчивых соединений и тем самым к снижению активности катализатора. В 1929 г. А. А. Баландиным была создана мультиплетная теория гетерогенного катализа, в основу которой положен принцип структурного и энергетического соответствия между катализатором и реагирующими веществами.
№24 Цепные реакции.Возможность цепного механизма химического взаимодействия была установлена H. А. Шиловым (1905). Теория цепных реакций создана трудами H. H. Семенова (СССР) и С. Хиншельвуда (Великобритания). Молекула a[u]b с ковалентной связью между атомами в активированном состоянии может распадаться на ионы:
в + В
(а)на атомы (или свободные радикалы):
AfTfIB —»- AfTl + ГПв
(б) 121В реакции (а) разрыв связи называется гетеролитическим или ионным, в реакции (б) — гомолитическим или радикальным.
При гомолитическом разрыве ковалентной связи образуются атомы или группы атомов (свободные радикалы) с повышенной реакционной активностью, обусловленной наличием неспарен-
ного электрона
. Цепная реакция всегда начинается гомолитическим разрывом связи в одной из реагирующих молекул, который происходит при поглощении энергии, и реакция продолжается самопроизвольно за счет возникновения новых реак-ционноспособных частиц в каждом акте процесса.
Существуют два типа цепных реакций: с неразветвленными и с разветвленными цепями. Примером реакции с неразветвленной цепью может служит фотохимический синтез хлорида водорода, который протекает со взрывом при облучении смеси водорода и хлора солнечным светом или светом горящего магния. Для цепных реакций характерны три стадии:
а) стадия зарождения цепи*
б) стадия развития цепи
в) стадия обрыва цепи (столкновение двух атомов)
№25 Необратимые и обратимые реакции. Химическое равновесие.
Когда при химическом взаимодействии хотя бы одно из исходных веществ расходуется полностью, реакцию считают необратимой,
123протекающей до конца. Примером необратимой реакции может быть разложение бертолетовой соли: KClO3-KCl + 1,5O2
К необратимым принято относить взаимодействия между веществами, в результате которых образуются осадки, газы и малодиссоциирующие вещества:
BaCl2+ Na2SO4 — BaSO4J + 2NaCl
Na2CO3 + 2HCl — CO2J + H2O + 2NaCI
NaOH + HCl — H2O + NaCl
Однако каждая из приведенных реакций лишь практически необратима. В химии растворов электролитов рассматривается возможность процессов
BaSO4-Ba21 +SOj-
CO2 + H2O — 2H+ + СО1~
H2O-H++ ОН"
обратных выделению осадка, газа и образованию малодиссоциирующего вещества.
Многие химические реакции протекают обратимо. Их особенность состоит в том, что они не идут до конца, в системе всегда остается (в большем или меньшем количестве) каждое из исходных веществ. К числу обратимых относятся, например, следующие взаимодействия:
СО + H2O ^ CO2 + H2
H2 + I2 ^ 2HI 3H2 + N2 ==± 2NH3
Реакцию, протекающую в правую сторону (-*•), называют прямой, а в левую (<—) —обратной. Если в системе скорость прямой реакции IT равна скорости обратной реакции 17, состояние системы называют химическим равновесием. Таким образом, кинетическим условием химического равновесия является равенство 7= JT.
№26. Растворы. Способы выражения концентрации растворов.
Раствор – жидкая, твердая или газ гомогенная система переменного состава образ 2 или > компонентами.
Газовые растворы. В газообразном состоянии частицы слабо взаимодействуют друг с другом, поэтому газовый раствор при обычном давлении можно считать смесью компонентов.
Жидкие растворы. К жидким растворам относят растворы газов, жидкостей и твердых веществ в жидких растворителях. В зависимости от природы растворителя различают водные и неводные растворы.
Твердые растворы — это фазы переменного состава, в которых атомы различных элементов расположены в общей кристаллической решетке, бее кристаллические твердые тела способны образовывать твердые растворы в большинстве случаев с узкими пределами концентрации растворяющегося компонента.
Способы выражения концентрации растворов. Концентрацией называют отношение количества (моль) или массы (г) вещества, содержащегося в растворе, к объему или массе раствора либо растворителя. Наиболее распространены следующие способы выражения концентрации растворов.
Молярная концентрация CМ — отношение количества вещества, содержащегося в растворе, к объему раствора [моль/м3]. При C=I моль/л раствор называют одномолярным, при C =0,01 моль/л — сантимолярным и т. п.

Моляльная доля N i =ni/(n1+n2+ni)
Молярная концентрация эквивалента (нормальная концентрация) CН - это отношение количества вещества эквивалента, содержащегося в растворе, к объему этого раствора [моль/м3].
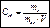
Эквивалентом называется реальная или условная частица вещества, которая может замещать, присоединять, высвобождать или быть каким-либо другим способом эквивалентна одному иону водорода в кислотно-основных или ионообменных реакциях или одному электрону в окислительно-восстановительных реакциях.
Моляльность С(X) —отношение количества вещества, содержащегося в растворе, к массе растворителя [моль/кг]. Например, 6(КОН/Н2О) = 2 моль/кг
Молярная доля N — отношение количества вещества одного из компонентов раствора к общему количеству вещества всех компонентов раствора. Титр раствора T — масса вещества, содержащаяся в одном миллилитре раствора [г/мл]. 
Массовая доля—отношение массы данного компонента в растворе к массе всего раствора.
З-н эквивалентов 

№27. Свойства разбавленных растворов не электролитов. 1-ый з-н Рауля.
1 з-н Рауля: отн. понижение давления нас. пара растворителя над раствором равно молярной доле растворенного вещества:
(pA0 - pA)/pA0 = nB (nA+nB), где pA0 и pA -давление нас. пара растворителя Анад чистым растворителем и над раствором соответственно; nА -число молей растворителя Ав растворе; nВ -число молей растворенного малолетучего неэлектролита Вв растворе.
№28. Кипение и кристаллизация растворов. 2-ой з-н Рауля.
Растворы кипят при t>> высоких и замерзают при t<< низких по сравнению с чистым растворителем.
∆tкип = kЭ * Cm ; ∆tкрист = kЭ*Cm — 2-ой з-н Рауля.
Повышение tкип р-ра или понижение тем-ры его кристаллизации прямопропорц. моляльной концетрации и не зависит от других факторов. 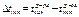 ,
,  ,
,
kЭ — эбуллиоскопическая и KK — криоскопическая константы.
Cm = 1000 * V2 / m1 = 1000 * m2 / M2 * m1
принципом Ле Шателье: если на систему, находящуюся в равновесии, оказывать внешнее воздействие, то равновесие смещается в том направлении, которое ослабляет эффект внешнего воздействия.
Смещение равновесия может быть вызвано изменением температуры, концентрации одного из реагентов, давления. Температура — тот параметр, от которого зависит величина константы равновесия химической реакции.
№29. Явление осмоса. Осмотическое давление. Закон Вант - Гоффа.
Раствор представлят собой однородную систему. Частицы растворенного в-ва и р-ля находятся в беспорядочном тепловом движении и равномерно распределяются по всему объему р-ра. Однако движение ч-ц растворенного в-ва и р-ля может стать направленным, если привести в соприкосновение два р-ра сразными концентрациями (С1 и С2). Молекулы р-ля и растворенного в-ва будут диффундировать преимущественно в том направлении, где концентрация их ниже. Так ,например если С1 > С2 , то молекулы р-ля с большей скоростью будут переходить в р-р сконц. С1, а молекулы растворенного в-ва- в р-р с конц. С1. Такая двустороняя диффузия приведет к С1=С2.
Диффузия может стать односторонней, еслир-ры разделить полупроницаемой перегородкой, пропускающей только молекулы р-ля. При условии, что С1>C2, молекулы р-ля с большей скоростью будут диффундировать в направлении С2®С1, и объем р-ра с концентрацией С1 несколько возрастет. Такая односторонняя диффузия р-ля через полупроницаемую перегородку наз. осмосом .Для колличественной хар-ки осмотических св-в р-ов по отношению к чистому р-лю вводиться понятие об осмотическом давлении, равном силе, приходящейся на единицу площади поверхности и заставляющей проникать молекулы р-ля через п/проницаемую перегородку. Величина осм. Давления зависит от t0 и С и не зависит от пироды р-ля и растворенного в-ва.
В 1886 г Вант – Гофф показал, что для разбавленных р-ов неэлектролитов зависимость осм. Давления от температуры и концентрации выражается: росм = CRT, где С-концентрация р-ра неэлектролита, моль/л.
№30. Растворы электролитов. Причина эл диссоциации. Сильные и слабые электролиты.
Электролитами наз вещества, диссоциирующие в воде, других полярных жидкостях или расплвах на ионы и способные проводить эл ток. Распад ве-ва на ионы называется эл диссоциацией. К электролитам относят соли, кислоты, основания.
Для электролитов Вант – Гофф ввел в уравнение попрвочный коэффициент i, называемый изотоническим коэффициентом и позволяющей использовать это ур-ние для любых р-ов:
 .
.
Эл диссоциация протекает самопроизвольно, т.е. энергия Гиббса системы понижается. Понижение энергии Гиббса системы обусловлено образованием сольватированных ионов. Энергия взаимодействия молекул р-ля с растворенным в-ом (энергия сольватации) достаточна,и чтобы разрушить химические связи в молекулах или ионных кристаллах.
Отношение числа молекул, диссоциированных на ионы, к общему числу молекул растворенного электролита наз степенью диссоциации a. По степени диссоциации в растворах все электролиты делятся на две группы. К первой группе относят эл-ты, степень диссоциации которых в растворах равна 1-це и почти не зависит от концентрации р-ра. Их называют сильными электролитами. К сильным электролитам в водных р-ах принадлежит большинство солей, щелочей, а также некоторые кислоты. Электролиты, где a<1 называют слабыми. К ним относят воду, ряд кислот, основания p-, d- и f-элементов. Между этими 2-мя группами не существует четкой границы, одно и то же в-во в одном р-ле проявляет св-ва сильного, а в другом –слабого электролита. Например: хлорид лития и иодид натрия.
№31. Св-ва разбавл р-ов сильных эл-ов. Изотонический коэфф. Связь и.к. и степени диссоц.
В растворах сильных эл-ов вследствии полной их диссоциации конц ионов велика. Поэтому св-ва таких р-ов будут сильно зависеть от степени взаимодействия входищих в их состав ионов как друг с другом, так и с полярными молекулами р-ля. Взаимодействие ионов в р-рах сильных эл-ов будет приводить к тому, что катионы и анионы будут испытывать взаимное притяжение, а ионы одного знака заряда будут испытывать отталкивание друг от друга. Поэтому в р-ре каждый произвольно выбранный ион будет окружен в среднем во времени, преимущественно противоположно заряженными ионами, как, например, в ионных кристалах.
№32. Слабые электролиты. Закон разбавления Освальда.
В р-ах слабых эл-ов процесс пртекает обратимо и, следовательно, к нему может быть применензакон действующих масс. Так , для процесса диссоциации уксусной кислоты
CH3COOHÛ CH3COO— + H+, константа равновесия KC будет равна:  . Константу равновесия для процесса диссоциации наз константой диссоциации КД. Она завасит от природы диссоциирующего в-ва и р-ля, а также от тем-ры, и не зависит от концентрации р-ра. При Т↑ КД обычно ↓.
. Константу равновесия для процесса диссоциации наз константой диссоциации КД. Она завасит от природы диссоциирующего в-ва и р-ля, а также от тем-ры, и не зависит от концентрации р-ра. При Т↑ КД обычно ↓.
В соответствии с принципом Ле Шателье подобная тем-рная зависимость константы КД указывает на то, что процесс диссоциации – экзотермический, т.е. суммарная теплота гидратации ионов выше энергии внутримолекулярных связей. КД указывает на прочность молекул эл-ов в данном р-ре. Чем меньше КД в данном р-ле, тем слабее диссоциирует электролит и тем, следовательно устойчивее его молекулы.
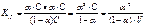 , где V=1/C-разведение р-ра.
, где V=1/C-разведение р-ра.
Это ур-ние известно как закон разведения Оствальда. В настоящее время за нуль принято считать потенциал стандартного водородного электрода. Такой электрод состоит из платинированной платины, контактирующей с газообразным водородом, находящимся под давлением 101кПа, и раствором, в котором активность ионов Н+ равна 1. Водородный электрод относится к газовым электродам, т.е. электродам, в котором по крайней мере один из реагентов является газообразным. Абсолютное значение потенциала водородного электрода неизвестно, но условно считают за нуль.
№33. Ионные реакции и их уравнения. Гидролиз солей.
В то же время для протекания любой хим реакции необходимо столкновение реагирующих частиц, т.е. сближение их до расстояния, на котором возможно электронные переходы. А так как скорость движения ионов в р-ре значительно превышает скорость движения молекул, то иенно ионы определяют реакциооную способность растворов не только сильных, но и слабых электролитов. Т. о.,и химические св-ва р-ра электролита складываются из св-в образующих его ионов
Гидролизом солей наз реакции обмена между водой и растворенными в ней солями. В результате протекания процесса гидролизасоли в растворе появляется некоторое избыточное кол-во ионов H+ или OH—, сообщающее раствору кислотные или щелочные св-ва. Пример: (процесс нейтрализации между соляной кислотой и гидроксидом натрия),
H+ +Cl— + Na+ + OH—®H2O+Na+ +Cl—. Равновесие процесса нейтрализации будет смещено в сторону образования наиболее слабого эл-та.
Показателем глубины протекания процесса гидролиза служит степень гидролиза b, представляющая собой отношение концентрации гидролизованных молекул СГИДР к исходной концентрации растворенных молекул эл-та:  .
.
Гидролиз принадлежит к числу обратимых процессов, поэтому положение его равновесия может быть смещено в ту или иную сторону изменением концентрации в-в—участников р-ии, а также тем-ры р-ра.
№34. Диссоциация воды. Ионное произведение воды. Водородный показатель.
Очищенная от посторонних примесей вода обладает незначительной электропроводностью, заметно возрастающей с повыщением тем-ры.H2O является слабым электролитом (H2OÛH+ +OH—).  . Концентрацию молекул воды можно расчитать,разделив массу 1л воды на массу ее моля: 1000/18=55.5 моль/л. Считая эту величину постоянной, можно записать СН+ СОН—=КД55,5=Кw.Константу равновесия Кw наз ионным произведением воды. Она сильно зависит от температуры.
. Концентрацию молекул воды можно расчитать,разделив массу 1л воды на массу ее моля: 1000/18=55.5 моль/л. Считая эту величину постоянной, можно записать СН+ СОН—=КД55,5=Кw.Константу равновесия Кw наз ионным произведением воды. Она сильно зависит от температуры.
В соответствии с теорией эл диссоциации ионы Н+ являются носителями кислотных св-в, а ионы ОН—-носителями основных св-в. поэтому раствор будет нейтральным, когда  ; кислым, когда aH>aOH; и щелочным, когда aH<aOH. Для хар-ки кислотности(щелочности) средывведен специальный параметр-водородный показатель или рН. Водородным показателем, или рН, называется взятый с обратным знаком десятичный логарифм активности водородных ионов в растворе: pH= -lg(aH+). При рН=7 среда нейтральная, рН>7- щелочная, pH<7- кислая.
; кислым, когда aH>aOH; и щелочным, когда aH<aOH. Для хар-ки кислотности(щелочности) средывведен специальный параметр-водородный показатель или рН. Водородным показателем, или рН, называется взятый с обратным знаком десятичный логарифм активности водородных ионов в растворе: pH= -lg(aH+). При рН=7 среда нейтральная, рН>7- щелочная, pH<7- кислая.
№35. Понятие об электродном потенциале. Равновесный э.п.. Устройство водородного потенциала. Стандартный электродный потенциал.
При погружении Ме в р-р начинается сложное взаимодействие Ме с компонентами р-ра. В результате взаимодействия происходит окисление Ме и его гидратированные ионы переходят в р-р, оставляя в Ме электроны, заряд которых не скомпенсирован положительно заряженными ионами в Ме: Ме +mH2O→Me(H2O)mn+ +ne—. Ме становится заряженным отрицательно, а р-р-положительно. Положительно заряженные ионы из р-ра притягиваются к отрицательно заряженной поверхности Ме. На границе Ме- раствор возникает двойной эл. слой. Между Ме и р-ом возникает разность потенциалов, которая наз электродным потенциалом или потенциалом электрода. По мере перехода ионов в р-р растет отрицательный заряд поверхности Ме и положительный заряд р-ра, что препятствует окислению Ме. Наряду с этой реакцией протекает обратная реакция-восстановление ионов Ме до атомов:
Me(H2O)mn+ +ne—→ Ме +mH2O. С увеличением скачка потенциала между электродом и р-ом скорость прямого процесса будет равна скорости обратного процесса, устанавливается равновесие: Ме +mH2O Û Me(H2O)mn+ +ne—. Равновесие имеет динамический хар-ер, процессы при равновесии идут с одинаковой скоростью в прямом и обратном направлениях. Потенциал, устанавливающийся в условиях равновесия электродной р-ии, наз равновесным электродным потенциалом
№36. Ряд напрежений металлов и его следствия.
Стандартные э.п. металлов указывают на меру восстановительной способности атомов металла и меру окислительной способности ионов металла. Чем более отрицательное значение имеет потенциал металла, тем более сильными восстановительными способностями обладает этот металл.
- - - - - -
Металлы обладают рядом общих свойств.
К общим физическим свойствам металлов относятся их высокая электрическая проводимость и теплопроводность, пластичность, т.е. способность подвергаться деформации при обычных и при повышенных температурах, не разрушаясь. Пластичность металлов имеет очень большое практическое значение. Благодаря этому свойству металлы поддаются ковке, прокатке, вытягиванию в проволоку (волочению), штамповке. Металлам присущи так же металлический блеск, обусловленный их способностью хорошо отражать свет, и непрозрачность.
В химическом отношении все металлы характеризуются сравнительной лёгкостью отдачи валентных электронов и, как следствие этого, способностью образовывать положительно заряженные ионы и проявлять в своих соединениях только положительную степень окисления. Многие металлы, например железо, хром, марганец, имеют в различных соединениях разную степень окисления, но она всегда положительна. В связи с этим металлы в свободном состоянии являются восстановителями. Восстановительная способность разных металлов неодинакова. Для реакций в водных растворах она определятся положением металла в ряду напряжений и концентрацией (активностью) его ионов в растворе.